Contenido del artículo
V PORFIRIAS
- Definición y clasificación
- Epidemiología
- Fisiopatología y defectos genéticos en la porfiria
- Porfirias hepáticas
- PORFIRIAS AGUDAS
- Porfiria aguda intermitente
- Porfiria abigarrada
- Coproporfiria hereditaria
- Deficiencia de deshidratasa de AAL (Plumboporfiria)
- Porfirias dobles
- PORFIRIA CUTANEA TARDA
- Porfirias eritropoyéticas
- Información adicional
DR. MARK G. PERLROTH
Definición y clasificación
Las porfirias son un grupo de enfermedades caracterizadas por la sobreproducción de compuestos porfirínicos y de sus precursores. En los animales la síntesis de porfirinas es necesaria para la producción del grupo hem, componente de la hemoglobina, citocromos (incluyendo el P450), catalasas, peroxidasas y otras enzimas oxidativas relacionadas con el metabolismo de los fármacos. Así como las porfirinas hem, que contienen hierro, catalizan la fosforilación oxidativa en animales a través de la acción de citocromos mitocondriales, las porfirinas clorofílicas, que contienen magnesio, catalizan las fotosíntesis en las plantas, liberando oxígeno a la atmósfera. Poéticamente, según lo describió Hans Fischer, ganador del premio Nobel, las porfirinas son sustancias que hacen que la sangre sea roja y el pasto verde.1
Epidemiología
Aunque los defectos genéticos y enzimáticos específicos existen durante toda la vida, la mayoría de las personas heterocigotas nunca presentan síntomas de la enfermedad.2 Cuando se desarrollan síntomas suelen aparecer después de la pubertad. Las formas agudas de la enfermedad se caracterizan por periodos latentes prolongados interrumpidos por ataques agudos. Durante la época latente la excreción de porfirina y precursor de porfirina en la orina varía de mínima a importante. Cuando se presentan síntomas las pruebas bioquímicas para estos productos son típicamente positivas.
Fisiopatología y defectos genéticos en la porfiria
Las porfirias se entienden mejor mediante el estudio del esquema básico de la síntesis del hem [ver figura 1]. El control del ritmo de síntesis depende de la enzima inicial, la ácido delta-aminolevulínico (AAL) sintetasa, dentro de las mitocondrias. En el citoplasma celular los anillos tetrapirrólicos permanecen en estado reducido, pero el número de residuos carboxílicos por anillo disminuye progresivamente de ocho a dos. Las tres últimas reacciones enzimáticas se llevan a cabo dentro de las mitocondrias, dando lugar finalmente a la producción del hem, que ejerce una acción represiva sobre la producción de la AAL sintetasa, que es la enzima inicial y que limita la velocidad en la vía metabólica. La pérdida de los grupos carboxilo hace que cada compuesto sucesivo sea menos hidrosoluble.
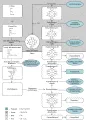
|
| Figura 1 |
| Síntesis del grupo hem y porfirias |
La conversión de protoporfirinógeno a protoporfirina mediante la pérdida de seis moléculas de hidrógeno da como resultado una serie de dobles enlaces alternos. La molécula con esta configuración absorbe la luz ultravioleta con una longitud de onda de 400 nm (banda de Soret), que es la responsable de la fluorescencia característica de todas las porfirinas. Los porfirinógenos intermedios se oxidan espontáneamente, en especial en presencia de luz. Las porfirinas resultantes se pierden de la vía sintética del hem y se excretan por la orina o por las heces, dependiendo de su solubilidad relativa en el agua.
Cada alteración específica en el patrón de excreción de las porfirinas y sus precursores es causada por una disminución en la concentración de alguna enzima de la vía de síntesis del hem. Pueden ocurrir deficiencias en cualquiera de las enzimas, desde la AAL dehidratasa hasta la ferroquelatasa. Debido a que los lugares de mayor producción de hem son la médula ósea (eritrocitos) y el hígado, las porfirias pueden ser agrupadas convenientemente en eritropoyéticas, hepáticas o mixtas, dependiendo del sitio primario de afección. Por sus manifestaciones clínicas tan claras, la facilidad para observar sus indicadores químicos en orina y su modo familiar de herencia, las porfirias se encuentran entre las primeras enfermedades que fueron definidas como errores innatos del metabolismo.
Con excepción de la porfiria eritropoyética congénita (PEC) y la deficiencia de AAL dehidratasa (plumboporfiria), las porfirias se heredan con patrón autosómico dominante. Dos padres que son heterocigotos para el mismo defecto enzimático pueden tener descendientes con formas mucho más graves de la enfermedad, mientras que los que son heterocigotos para diferentes deficiencias enzimáticas pueden tener hijos con porfiria mixta o dual.
Entre los adelantos en biología molecular de la década pasada se encuentran el mapeo de las localizaciones cromosómicas y la secuencia de los genes que codifican las enzimas que participan en la biosíntesis de la porfirina,1 así como la identificación de muchas mutaciones específicas. Estas pueden incluir mutaciones puntiformes, inserciones o deleciones que cambian la secuencia de aminoácidos de una enzima y por lo tanto interfieren con su capacidad para fijar o liberar intermediarios o con su estabilidad.3
El mejor conocimiento de la biología molecular de la síntesis de la porfirina no solo ha permitido adelantar en la evaluación clínica y el diagnóstico, sino también en el uso de un tratamiento eficaz con hem para las porfirias agudas. El tratamiento con molécula hem reprime la actividad de la enzima hepática AAL sintetasa, disminuyendo así la producción excesiva de precursores de porfirina en el hígado, con el alivio consecuente de los síntomas de las porfirias agudas.
Porfirias hepáticas
PORFIRIAS AGUDAS
Las porfirias agudas son, en orden de prevalencia en Norteamérica, la porfiria aguda intermitente (PAI), la porfiria abigarrada (PA), la coproporfiria hereditaria (CPH) y la deficiencia de AAL deshidratasa. Como grupo se caracterizan por la presencia de ataques neuroviscerales (manifestaciones neurológicas y dolor abdominal), y por varias de cinco "P" (en inglés, n. del t.): (1) inicio después de la pubertad, (2) alteraciones psiquiátricas, (3) dolor, (4) polineuropatía y (5) fotosensibilidad (que se encuentra solo en la CPH y la PA). En su periodo de latencia los signos y síntomas pueden estar ausentes; sin embargo, los periodos agudos se pueden precipitar por las cuatro "M" (en inglés, n. del t.): (1) medicamentos (incluyendo estrógenos y alcohol) [ver tabla 1], (2) menstruación y periodo premenstrual, (3) desnutrición (ingestión escasa de carbohidratos o periodos de ayuno prolongados), y (4) enfermedades médicas (y cirugías). Aunque la PCT también es causada por una deficiencia enzimática en el hígado, solo causa fotosensibilidad y se analiza en forma separada [ver adelante, Porfiria cutánea tarda].
|
|||||||||
Nota: ver también la página de la American Porphyria Foundation http:/www.enterprise.net/apf. La American Porphyria Foundation cobra una canttidad por entrar a su dirección.
|
|||||||||
La aparente contradicción entre un defecto genético establecido con un patrón clínico intermitente se explica por el hecho de que la AAL sintetasa hepática, que en condiciones normales es la enzima limitante en la síntesis del hem, es inducida por ciertos estímulos endógenos (v.gr., estrógenos) y exógenos (v.gr., ayuno o drogas), precipitando los ataques clínicos agudos de la enfermedad. El defecto básico en las diversas porfirias es la deficiencia parcial de una enzima no limitante. Este defecto puede ser bien tolerado en periodos en los que la producción del grupo hem sea normal en el hígado; sin embargo, cuando se induce la producción de AAL sintetasa, hay un aumento en la síntesis de precursores que sobrepasan a la capacidad del paso enzimático deficiente en la vía de síntesis, acumulándose los precursores próximos al bloqueo enzimático, que posteriormente salen del hepatocito hacia la circulación. Dependiendo de la enzima afectada y de la magnitud de inducción de AAL sintetasa, el exceso de precursores puede consistir de AAL y porfobilinógeno (PBG) en combinación con cantidades variables de uroporfirinógeno, coproporfirinógeno y protoporfirinógeno. Ya que los porfirinógenos se oxidan fácilmente hacia su respectiva porfirina fluorescente, por lo general se puede medir la excreción de AAL, PBG y porfirinas. La descarboxilación progresiva de los productos intermedios de porfirinógeno en la vía sintética del hem causa una pérdida de solubilidad en agua; por lo tanto, la coproporfirina y la protoporfirina se excretan menos por el riñón y más por el hígado, siendo necesario el análisis de heces para su identificación.
Al parecer, los factores precipitantes actúan mediante la inducción de la AAL sintetasa hepática, ya sea por estimulación directa de la enzima o por falta de control de la síntesis de AAL sintetasa secundaria a la disminución del hem disponible. La fijación del hem por proteínas intracelulares podría ser una manera de reducir este producto. La disminución de la ingesta de carbohidratos aumenta la actividad de la AAL sintetasa, pero el mecanismo de este fenómeno se desconoce. La mayor frecuencia de las porfirias hepáticas en mujeres, su aparición después de la pubertad y los fenómenos de exacerbación en la época premenstrual y menstrual sugieren la participación de los estrógenos y la progesterona en el curso de la enfermedad.
Es probable que existan lesiones adquiridas en la producción de porfirinas. El plomo inhibe a la AAL deshidratasa, lo que ocasiona aumento en la excreción urinaria de AAL, pero no de PBG. Ocurren dolor abdominal, neuropatía periférica y otros datos de porfiria aguda. Puede ocurrir una lesión bioquímica semejante en la tirosinemia hereditaria secundaria a acúmulo de succinilcolina, un potente inhibidor de la AAL deshidratasa. Las enfermedades crónicas del hígado, la hemódiálisis y las neoplasias están relacionadas con un aumento en la excreción de coproporfirinas.
Porfiria aguda intermitente
El patrón autosómico dominante de la porfiria aguda intermitente (PAI) se asocia con una disminución de cerca del 50 por ciento de la actividad de la enzima deaminasa de PBG. Se han identificado más de 100 mutaciones diferentes que afectan la estabilidad de la actividad catalítica de la deaminasa de PBG, así como 10 polimorfismos genéticos neutrales.4
La prevalencia del defecto genético en la PAI varía de uno en 500 en Escandinavia a uno en 1,500 en Francia. Estudios de familias demostraron que el 80 a 90 por ciento de los casos son latentes. Con base en estas cifras, la incidencia esperada de episodios agudos debe ser de uno a dos por 15,000, mucho más alto que la incidencia reportada de uno a dos por 100,000. Esta discrepancia sugiere que muchas mutaciones se expresan en forma leve o que muchos casos no se diagnostican de modo correcto.4
En estudios de 92 familias afectadas, 89 tenían disminución en la actividad de la deaminasa de PGB causada por reducción en las propiedades cinéticas y estabilidad de la enzima, mayor fijación o liberación defectuosa del sustrato. Las tres familias restantes tenían niveles normales de deaminasa de PBG en los eritrocitos.3 Una posible explicación para esta discrepancia es que tanto la PBG deaminasa de los hepatocitos y eritrocitos deriva de un gen que contiene 15 exones, pero el ARN mensajero (ARNm) se fragmenta en forma diferente en los dos tejidos, lo que causa que la regulación de la síntesis del hem sea distinta.5 Mientras que la enzima hepática se traduce de los exones 1 y 3 al 15, el proceso eritrocítico elimina el exón 1, traduciendo la enzima de los exones 2 al 15. En el hepatocito, una mutación en el primer intrón inmediatamente proximal al sitio de ruptura evita la transcripción del exón 1, alterando la enzima hepática y reduciendo la actividad de la deaminasa de PBG en un 50 por ciento. En el eritrocito la transcripción comienza en el exón 2 y no tiene efectos adversos sobre la expresión de la enzima. Por el contrario, puede ocurrir deficiencia aislada de deaminasa de PBG eritrocitaria en personas con función enzimática normal en el hígado, dando . resultados falsos positivos.6
Aunque el defecto en la deaminasa de PBG no tiene importancia en situaciones normales en los pacientes con PAI, la inducción de la AAL sintetasa por cualquiera de los cuatro factores "M" produce acúmulo de precursores AAL y PBG, que son hidrosolubles y se excretan en grandes cantidades en la orina. El AAL es incoloro, pero el PBG se condensa en polímeros café-rojizo (porfobilina), que suelen ser visibles en la orina durante los ataques agudos. La condensación espontánea de PBG, así como cierta conversión a uroporfirina en los tejidos extrahepáticos explica los niveles elevados de porfirina en la orina. En la PAI la producción de porfirinógeno, y en consecuencia de porfirina, es poca, por lo que no ocurre fotosensibilidad.
Las manifestaciones clínicas y características diagnósticas de la PAI son variadas [ver tabla 2]. Las manifestaciones de neurotoxicidad a nivel cortical incluyen cambios de carácter, irritabilidad y psicosis franca. Las lesiones hipotalámicas pueden ocasionar el síndrome de secreción inapropiada de hormona antidiurética (SIHAD) con hiponatremia, que puede causar crisis convulsivas y trastornos psiquiátricos. La disfunción de los nervios craneales, la paresia bulbar y la dificultad en la deglución pueden causar neumonía por aspiración. La neuropatía periférica produce dolor y paresia motora. Si los nervios intercostales y frénicos se afectan puede ocurrir insuficiencia ventilatoria severa; la afección de los nervios autosómicos pueda ocasionar dolor abdominal, íleo paralítico, taquicardia, hipertensión arterial y, con menos frecuencia, diarrea. Debido a que los signos y síntomas son de origen neuropático, no suelen existir (o son leves) fiebre, leucocitosis y otros signos de inflamación.
|
||||
* Extremidades, articulaciones, espalda o cabeza. |
La PAI debe ser incluida en el diagnóstico diferencial del dolor abdominal de causa desconocida, en los trastornos psiquiátricos agudos y en las polineuropatías agudas. Son de ayuda para el diagnóstico los antecedentes familiares positivos, la presencia de dolor en la espalda y extremidades, así como en el abdomen, y el desarrollo agudo de hipertensión arterial durante el ataque. La sospecha será mayor cuando los síntomas ocurran en la fecha esperada de la menstruación, después de la toma de ciertas drogas (como alcohol o anestésicos) [ver tabla 1], durante un régimen dietético o después de una cirugía.
Puede investigarse PBG en forma rápida en una muestra de orina por medio de la prueba de Watson-Schwartz o de Hoesch; en ambas se utiliza el reactivo de Ehrlich como cromógeno. En cualquier caso, se deberá recolectar la orina de 24 horas en recipientes opacos y refrigerarse para entregarla a un laboratorio calificado para el análisis cuantitativo de AAL, PBG y porfirinas.
Debido que otras porfirias hepáticas que pueden ser inducibles (como la deficiencia de deshidratasa de AAL, la CPH y la PA) pueden producir síndromes neuropáticos idénticos sin lesiones por fotosensibilidad y caracterizarse por excreción excesiva de PBG, AAL o ambos, el diagnóstico definitivo tendrá que realizarse mediante el análisis en las heces y la prueba de deaminasa de PBG en los eritrocitos. Esta última prueba tiene una sensibilidad de 80 a 90 por ciento y es confiable durante el periodo latente de la enfermedad, cuando pueden estar ausentes las alteraciones bioquímicas urinarias. También es útil para la detección de los familiares asintomáticos del enfermo, que pueden padecer la forma latente, aunque puede estar falsamente elevada (i.e., normal) si la cuenta de reticulocitos es alta.3 Si se ha identificado una sola mutación en la descendencia, los familiares pueden ser estudiados usando técnicas modernas de análisis de ADN.
La identificación temprana de la etiología de los síntomas porfíricos evitará la realización de pruebas innecesarias y permitirá el tratamiento más temprano. Puede recomendarse el escrutinio familiar, aun cuando éste puede producir más ansiedad y mayor dificultad para obtener un seguro de salud o empleo. Es muy importante evitar la ingestión de los medicamentos conocidos como desencadenantes de un ataque agudo [ver tabla 1]. También se recomienda una dieta isocalórica con contenido elevado de carbohidratos, así como consejo genético.
El tratamiento de apoyo consiste en alivio del dolor con meperidina o fenotiacinas e infusión de glucosa a una velocidad de 10 a 20 g/hr (400 a 500 g/día). Si aparece hiperglucemia excesiva se pueden agregar pequeñas dosis de insulina. Se han recomendado los agentes beta bloqueadores para el tratamiento de la taquicardia y la hipertensión arterial. Se ha usado gabepentin en forma segura para el control de las crisis convulsivas.7 Si no ocurre mejoría sintomática rápida en 24 horas o si existe neuropatía motora debe iniciarse de inmediato tratamiento con hem y medirse la capacidad vital a intervalos frecuentes. El peligro de muerte por insuficiencia respiratoria rápidamente progresiva es grande, por lo que debe disponerse del equipo necesario y personal capacitado para la intubación orotraqueal y la asistencia ventilatoria. Será necesario el manejo adecuado de soluciones parenterales para evitar la hiponatremia. Puede requerirse intubación nasogástrica o nutrición parenteral si los síntomas bulbares dificultan la deglución.
El uso de hematina, forma férrica del grupo hem, para aquellos individuos cuyos síntomas agudos no responden con rapidez a las medidas ya señaladas, es la medida más importante en el tratamiento de estos enfermos.8 Debido a que el hem y la hematina son potentes inhibidores de la AAL sintetasa hepática y como esta enzima tiene una vida media muy corta, los niveles plasmáticos y urinarios de AAL y PBG pueden normalizarse en uno o dos días, lo que permite una rápida mejoría de los síntomas [ver figura 2] Las neuropatías periféricas establecidas no revierten de inmediato, pero se detiene su progresión. La dosis de hematina es de 1 a 4 mg/kg de peso disueltos en solución salina fisiológica y administrados por vía intravenosa durante 20 minutos cada 12 a 24 horas.8 Suele observarse mejoría en 2 a 7 días. Se ha publicado que en algunos casos la infusión de hematina se relaciona con una coagulopatía caracterizada por prolongación del tiempo parcial de tromboplastina, del tiempo de protrombina y de la fibrinolisis.8 En la actualidad se dispone en Europa de arginato de hem, pero aún no en los Estados Unidos. Esta es una preparación de hem mucho más estable, con excelente eficacia y que causa menos o ningún efecto trombótico.

|
| Figura 2 |
| Porfiria aguda intermitente, respuesta a tratamiento |
La hematina también es útil cuando se administra en forma profiláctica cada semana a mujeres con exacerbación menstrual de la PAI. En un estudio se redujo la excreción de precursores de porfirina a casi lo normal y los síntomas desaparecieron.8
Aunque los anticonceptivos orales y los estrógenos aumentan la excreción de AAL y PBG y pueden precipitar los ataques agudos, de manera paradójica también pueden ser útiles en algunas mujeres mediante la inhibición de los ciclos menstruales y los ataques agudos que pueden ocurrir en las etapas menstruales. Aunque puede aparecer PAI durante el embarazo, los episodios sintomáticos son raros. La profilaxis y el tratamiento son los mismos ya descritos.
El pronóstico de los pacientes con PAI es probablemente mejor que lo que comúmente sugiere la literatura médica. Con el advenimiento de la prueba de laboratorio para deanimasa de PBG en los eritrocitos y el análisis del ADN, es claro que existen muchos más pacientes con PAI de lo se había supuesto mediante la evaluación clínica. El evitar la realización de cirugías innecesarias o los medicamentos nocivos [ver tabla 1] disminuye el riesgo de los ataques agudos. Si éstos ocurren, los métodos cada vez más sofisticados y actualmente disponibles para el cuidado intensivo de pacientes con insuficiencia respiratoria y neumonía pueden aumentar las posibilidades de supervivencia. Finalmente, la utilización pronta de la hematina suspenderá los ataques agudos antes de que se establezca la parálisis. Cuando se requiere apoyo total y asistencia respiratoria continua, el tratamiento con hematina es menos eficaz y el pronóstico es malo.
Porfiria abigarrada
La porfiria abigarrada, también conocida como porfiria sudafricana porque es relativamente común entre la población blanca de ese país, es más rara en los Estados Unidos que la PAI. Se atribuye a disminución del 50 por ciento en la actividad enzimática de la protoporfirinógeno oxidasa (PPO) y se relaciona con una sobreprodución persistente de protoporfirinógeno, coproporfirinógeno y uroporfirinógeno (en orden cuantitativo descendiente) y por aumento en la excreción de las porfirinas correspondientes. La protoporfirina, siendo la menos hidrofílica, es excretada por el hígado y se reabsorbe en forma parcial en el intestino. Debe de ser buscada en las heces, en donde se encuentra elevada en el 95 por ciento de los casos. Los ensayos para protoporfirina plasmática pueden ser aún más sensibles.10 La coproporfirina se encuentra en heces y orina. Durante los ataques agudos el AAL y el PBG se excretan en cantidades anormales en la orina y la prueba de Watson-Schwartz puede ser positiva; durante la fase de remisión la excreción de AAL y PBG puede ser normal. El aumento en la excreción de estos metabolitos intermedios refleja la inducción de la enzima AAL sintetasa. Ya que la enzima deficiente, la PPO, se encuentra situada dentro de la mitocondria, los eritrocitos maduros no pueden ser utilizados para la realización de los estudios definitivos. Se ha estudiado a los normoblastos, los leucocitos, el músculo esquelético y otros tejidos para definir las alteraciones bioquímicas. En la actualidad se ha clonado ya el gen de esta enzima en el humano.10
La presencia de niveles circulantes aumentados de porfirinas predispone a estos pacientes a sufrir lesiones dérmicas. Típicamente las lesiones aparecen en las áreas expuestas al sol; se pueden encontrar ampollas, así como úlceras superficiales en diferentes etapas de curación y cicatrices. La fragilidad mecánica es común, especialmente en los lugares de protuberancias óseas como los nudillos y tobillos. Existe también hipertricosis moderada en la cara y sienes. Los enfermos con PA pueden desarrollar lesiones por fotosensibilidad hasta en interiores, porque la banda de Soret (con longitud de onda de 400 a 410 nm) puede penetrar los cristales de las ventanas ordinarias y las pantallas solares. Las lesiones dérmicas o neuropáticas pueden ocurrir juntas o por separado; los miembros de una familia pueden tener una o ambas lesiones. Las lesiones dérmicas pueden ser idénticas a las observadas en la coproporfiria hereditaria y en la porfiria cutánea tarda, por lo que es necesario realizar diagnóstico diferencial con estos padecimientos. La presencia de lesiones neuropáticas sin manifestaciones cutáneas obliga a descartar PAI. Cuando ocurren lesiones cutáneas y neuropáticas en un mismo individuo o familia, deben considerarse en el diagnóstico diferencial la coproporfiria hereditaria o una de las porfirias dobles.
La diferenciación bioquímica se basa en el hecho que la PA se debe a un bloqueo enzimático distal a la síntesis de protoporfirinógeno hepático. Por lo tanto, se encuentra un exceso de protoporfirina en las heces. Por el contrario, la PAI y la coproporfiria hereditaria son causadas por la disminución enzimática proximal al protoporfirinógeno, y se relacionan con niveles normales o casi normales de protoporfirina en las heces. Debido a que las porfirinas de las heces que producen fluorescencia pueden provenir de protoporfirina derivada de grupos hem, se deberá evitar la ingesta en la dieta de carnes rojas durante la recolección de las muestras fecales y realizar en forma simultánea la prueba de sangre oculta en heces para evitar la confusión con protoporfirina derivada del hem.
Los factores precipitantes, los principios de profilaxis y el tratamiento son los mismos que para la PAI. La ausencia de una prueba fácilmente disponible para detectar el defecto enzimático en la PA hace necesaria la obtención de muestras de orina y de materia fecal de los miembros de la familia para ofrecer consejo genético. Los resultados negativos de estos estudios no deben ser considerados definitivos a menos que los miembros de la familia hayan alcanzado la pubertad. El tratamiento de las lesiones dérmicas se discute en otra parte del capítulo [ver adelante, Porfirias eritropoyéticas].
Coproporfiria hereditaria
La CPH es una porfiria hepática aún menos común; su patrón hereditario es autosómico dominante, y puede atribuirse a una actividad insuficiente de la enzima coproporfirinógeno oxidasa, causando producción excesiva de coproporfirina (principalmente coproporfirina III) y sus precursores. Al igual que en la PAI y la PA, la CPH se exacerba química y clínicamente por la inducción de AAL sintetasa. Se ha clonado el gen y se han aislado 10 mutaciones, así como varios polimorfismos neutrales.11 Dos casos fueron homocigotos desde el punto de vista fenotípico (actividad de la enzima del 10 por ciento contra 50 por ciento en los casos habituales).12
Cuando ocurren los síntomas se encuentran aumentados los niveles de AAL y PBG en la orina, con prueba positiva de Watson-Schwartz. Los niveles de coproporfirina en orina y heces están aumentados. Se pueden encontrar también pequeñas cantidades de protoporfirina en heces (aunque son más altos los niveles de coproporfirina), pero considerablemente menores que las que se encuentran en la PA. El aumento discreto en los niveles de coproporfirina urinaria como hallazgo único es inespecífico y se puede encontrar en trastornos hepáticos, hematológicos y tóxicos.
El espectro sintomático es indistinguible de la PA, y los síntomas son muy parecidos al dolor y neuropatía de la PAI y a las lesiones dérmicas que se observan en la porfiria cutánea tarda. La disminución de la actividad enzimática de la coproporfirinógeno oxidasa puede ser detectada en los linfocitos por laboratorios especializados; los valores se encuentran generalmente al 50 por ciento de lo normal en pacientes mayores de un año.
El tratamiento y la profilaxis de la CPH son los mismos que para la PA y la PAI.
La respuesta al tratamiento también es similar [ver tabla 1].
Deficiencia de deshidratasa de AAL (Plumboporfiria)
La forma menos frecuente de las porfirias hepáticas es causada por deficiencia parcial de la deshidratasa de AAL. Debido a que esta enzima, que convierte al AAL a PBG, también es inhibida por el plomo, se ha llamado a este padecimiento plumboporfiria. Solo se han reportado cuatro casos. El gen de la deshidratasa de AAL tiene dos exones promotores, uno es eritroide específico y el otro tiene un sitio ubicuo, en un arreglo análogo al de la deshidratasa de PBG, lo que permite el control independiente de la síntesis de hemoglobina por las células eritroides en respuesta a las necesidades hematológicas. En el hígado normal la deshidratasa de AAL se encuentra 80 a 100 veces más que la enzima que limita la velocidad, la AAL sintetasa, y una reducción del 50 por ciento (heterocigota) no es clínicamente aparente. Este padecimiento se hereda como un trastorno autosómico recesivo y se caracteriza por reducción en la actividad de deshidratasa de AAL a 1 al 2 por ciento de lo normal,13 se produce aumento en la excreción de AAL, pero no de PBG. Por lo tanto, a pesar de los síntomas neuroviscerales típicos (manifestaciones neurológicas y dolor abdominal), las pruebas habituales de escrutinio (Watson-Schwartz y Hoesch) son negativas y se requiere el análisis cuantitativo de los niveles urinarios de AAL en 24 horas. En el diagnóstico diferencial debe incluirse la intoxicación por plomo y la tirosinemia familiar. El tratamiento de la deficiencia de deshidratasa de AAL es el mismo que el de la PAI.
Porfirias dobles
Cuando se afecta más de una enzima en la secuencia biosintética en una sola generación, la persona puede tener una porfiria doble, que se caracteriza por un patrón mixto de excreción de porfirinas. En el caso de la llamada porfiria de Chester,14,15 los miembros de una familia de Chester, Inglaterra, tuvieron defectos tanto en la concentración de deaminasa de PBG (mapeada en el cromosoma 11) como en la concentración de protoforfirinógeno oxidasa (mapeada al cromosoma 14), que se manifestaron, respectivamente, como PAI y PA concomitantes. Aunque no ocurrió fotosensibilidad, existieron síntomas neuroviscerales. El patrón de excreción de porfirinas varió entre los diferentes miembros de la familia.
Existen reportes de otras familias con porfirias dobles, con manifestaciones cutáneas y neuroviscerales compatibles con PAI y PCT16 o PA y PCT concomitantes.17 Es probable que las porfirias dobles sean más frecuentes de lo que se ha pensado.
PORFIRIA CUTANEA TARDA
La PCT es la más común de las porfirias. A diferencia de otras porfirias hepáticas, la PCT es mucho más frecuente esporádica (tipo I) que familiar; es más común en hombres que en mujeres, y alrededor del 20 por ciento de los casos son familiares (tipo II).
La PCT es causada por la disminución parcial (> 50 por ciento) de la actividad enzimática de la uroporfirinógeno descarboxilasa (UPDC) hepática. En la PCT de tipo I la disfunción de la UPDC se limita al hígado. Las lesiones se deben a la producción y excreción excesivas de uroporfirina. Se ha desarrollado PCT esporádica después de la exposición a estrógenos o químicos exógenos; un gran brote de la enfermedad fue detectado después de la ingestión de granos contaminados con el fungicida hexaclorobenceno. La PCT también se ha relaciona con la hepatitis crónica, la hepatopatía alcohólica crónica, la sobrecarga de hierro y, en raros casos, con hepatoma. Recientemente ha surgido asociación con el virus de la hepatitis C. Se han encontrado anticuerpos contra el virus de la hepatitis C (VHC) en el 60 a 80 por ciento de los casos no familiares; sin embargo, esta asociación varía con la prevalencia local de la infección por VHC. La prevalencia de anticuerpos contra el virus de la hepatitis B (VHB) es mucho menor en las mismas poblaciones.18 La PCT tiene diversas características clínicas y diagnósticas [ver tabla 3]. Debido a que no se induce sintetasa de AAL, la excreción de AAL y PBG se encuentran en límites normales y la prueba de Watson-Schwartz es negativa. No se asocia con síntomas neuropáticos agudos.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
CFTH-capacidad total de fijación de hierro.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
En la forma familiar de la PCT (tipo II), heredada con patrón autosómico dominante, existe una reducción aproximada del 50 por ciento en la actividad de UPDC, tanto en los eritrocitos como en el hígado. Este hallazgo es compatible con la herencia de un gen defectuoso único. Se ha mapeado el gen de la UPDC y se han identificado 10 mutaciones.19 La expresión clínica es variable y la mayoría de los casos son latentes. La progenie de dos heterocigotos puede dar lugar a un individuo fenotípicamente homocigoto con manifestaciones graves que comienzan en la infancia temprana [ver adelante, Porfiria hepatoeritropoyética].
La presencia de niveles anormales de uroporfirinas en sangre y otros tejidos ocasiona fotosensibilidad, con las mismas lesiones dérmicas en áreas expuestas al sol que se observan en la PA y en la PCH y que inician en forma gradual. La enfermedad puede diferenciarse químicamente de la PA y de la PCH por el predominio de uroporfirina en la orina sobre la coproporfirina y la ausencia de una elevación notable de protoporfirina en las heces.
Se encuentra evidencia de exceso en las reservas de hierro en el 60 a 85 por ciento de los pacientes, que es severa en el 20 por ciento. Estudios recientes encontraron mutaciones relacionadas con hemocromatosis en el 44 y 73 por ciento de los pacientes con PCT esporádica (tipo I).20,21 Sin embargo, la PCT franca es poco común en la hemocromatosis. El hierro parece tener un papel central en esta enfermedad al causar la formación de radicales reactivos del oxígeno, que oxidan el uroporfirinógeno a uroporfirina y generan otros productos que inactivan en forma irreversible la UPDC. Los casos familiares (tipo II) con deficiencia genéticamente definida de UPDC son vulnerables al mismo mecanismo.
La base del tratamiento en todos los casos de PCT es la reducción del hierro, incluso en pacientes en quienes no se demuestra evidencia bioquímica de sobrecarga. Las flebotomías repetidas de una unidad de sangre dos veces al mes hasta un total de 5 a 10 L, dirigidas por el hematocrito del paciente, producirán disminución en la excreción de uroporfirina y en la fotosensibilidad. La mejoría ocurre después de varios meses hasta un año. Los pacientes que están en hemodiálisis o que tienen anemia deben recibir eritropoyetina. Para los pacientes que no toleran la flebotomía se han obtenido los mismos resultados con la infusión subcutánea lenta de desferroxamina mediante bombas portátiles (1.0 a 1.5 g en 8 a 10 ml de agua estéril durante 8 a 10 horas cada noche, durante 5 noches a la semana por un lapso de 2 a 5 meses).22 De igual importancia son las medidas para evitar la exposición al sol y la ingestión de alcohol, hierro y estrógenos.
La cloroquina, que forma complejos con la uroporfirina, ha causado mejoría en 3 a 6 meses. Puede ocurrir hepatotoxicidad. Después del tratamiento exitoso para la infección por VHC ha ocurrido resolución de la PCT tipo I.23 Puede intentarse el tratamiento de la fotosensibilidad con ß-carotenos, pero los resultados han sido contradictorios. [ver adelante, Porfirias eritropoyéticas], Debe vigilarse la excreción de uroporfirina en intervalos de 6 a 12 meses después de la remisión para evaluar la necesidad de nuevos cursos de tratamiento.
Porfirias eritropoyéticas
Dos porfirias son causadas por una alteración enzimática mayor del sistema eritropoyético: la porfiria eritropoyética congénita y la protoporfiria. (que también incluye un defecto en los hepatocitos) y la porfiria eritropoyética congénita. En comparación con las porfirinas de síntesis hepática, la síntesis de porfirinas en el sistema eritropoyético responde a cambios en la masa de eritrocitos y a los niveles de oxigenación de los tejidos; por lo tanto, la anemia, la hipoxia y la eritropoyetina son importantes reguladores de la producción del hem (y de la hemoglobina). Unicamente los reticulocitos y sus precursores en la médula ósea responden a estos estímulos. Los eritrocitos maduros carecen de mitocondrias y núcleo y, por lo tanto, no posee la maquinaria enzimática completa para la síntesis de porfirinas.
PORFIRIA ERITROPOYETICA CONGENITA
La PEC, también conocida como enfermedad de Günther, es extremadamente rara. Se hereda como un rasgo autosómico recesivo. El origen enzimático del trastorno parece ser un déficit severo (< 1 a 36 por ciento) en la uroporfirinógeno sintetasa III, enzima que cataliza la síntesis de la molécula asimétrica de uroporfirinógeno III a partir del tetrapirrol hidroximetilbilano lineal. Se ha secuencia el gen humano y se han identificado por lo menos 18 mutaciones.24
La CEP promueve el acúmulo de uroprofirinógeno I y de coproporfirinógeno I, isómeros que no son intermediarios en la síntesis del hem. La oxidación de estos productos hacia su respectiva porfirina, uroporfirina I y coproporfirina I comienza en la etapa fetal. El exceso de porfirinas ocasiona coloración de huesos y dientes (eritrodoncia), hemólisis, orina oscura y fotosensibilidad; todos estos datos se detectan desde la infancia. Los hallazgos más notables se relacionan con formación rápida de vesículas, cicatrización y mutilación, posteriores a la exposición a la luz solar. Los cambios tardíos pueden confundirse con esclerodermia. La alopecia, la conjuntivitis, la inflamación corneal y la queratomalacia son otras manifestaciones. En ocasiones existen deformidades óseas, atribuibles a los efectos tóxicos del depósito de porfirina en el hueso y quizá al raquitismo secundario a la no exposición a la luz solar. La anemia hemolítica es común y frecuentemente se acompaña de esplenomegalia; también puede existir hiperesplenismo con leucopenia y trombocitopenia. La severidad depende de la actividad residual y la estabilidad de la enzima mutada. La mayoría de los casos son heterocigotos compuestos.24
Puede demostrarse fluorescencia de los dientes, así como de los eritrocitos y sus precursores, cuando son irradiados con luz correspondiente a la banda de Soret. Al igual que en otras porfirias que producen fotosensibilidad, los cristales convencionales y los filtros ultravioleta son ineficaces para detener el espectro dañino de luz. Las transfusiones permiten la supresión de la eritropoyesis y, por lo tanto, de la síntesis del hem; cuando ocurre hemólisis, ésta mejora con la esplenectomía. Debe evitarse la exposición a la luz solar y aplicar cuidados locales a las lesiones dérmicas.
Los absorbentes orales, como la colestiramina y más recientemente el carbón, parecen disminuir la reabsorción de las porfirinas en el intestino.25 Incluso con el tratamiento, con frecuencia ocurre mutación progresiva y muerte prematura. El trasplante de médula ósea ha sido exitoso para revertir el defecto metabólico en cuatro pacientes.24 Se ha logrado la transducción in vitro de células hematopoyéticas progenitoras humanas de PEC, lo que sugiere que puede desarrollarse un tratamiento génico ex vivo.24,26
PORFIRIA HEPATOERITROPOYETICA
Como se mencionó antes, los pacientes con PCT congénita (tipo II) son heterocigotos típicos, y la condición suele ser latente. La descendencia homocigota ( o heterocigota compuesta) tiene deficiencia marcada de UPDC, manifestada por fotosensibilidad severa que comienza en la infancia temprana. Se han reportado alrededor de 30 casos.19 Las lesiones son causadas por acúmulo y depósito diseminado de uroporfirina III y simulan los causados por la PEC (ver antes). Los pacientes rara vez responden a la flebotomía o a cloroquina.
PROTOPORFIRIA ERITROPOYETICA
Debido a que la protoporfiria eritropoyética (PEP) se asocia con sobreproducción de protoporfirina por los etritocitos así como por las células hepáticas, se le ha llamado de otras dos formas: protoporfiria eritropoyética y porfiria eritrohepática. La enfermedad se hereda con un patrón autosómico dominante y se atribuye a la ausencia parcial de la enzima mitocondrial ferroquelatasa (hem sintetasa), la cual fija el Fe2+ a la protoporfirina IX. Se ha aislado el ADN complementario que codifica a la ferroquelatasa humana y solo se expresa una forma de la enzima tanto en los tejidos eritroides como no eritroides, a diferencia de la deaminasa de PBG. La relación entre la ferroquelatasa y la actividad de la sintetasa de AAL (paso que limita la velocidad) es de 2.6:1 en las células eritroides y de 500:1 en el hígado. Por lo tanto, la reducción de la ferroquelatasa se expresa al inicio y principalmente como un trastorno eritroide.27 La protoporfiria no se asocia con aumento urinario en la excreción de porfirinas, AAL o PBG. Con frecuencia, aunque no siempre, aumentan los niveles de protoporfirina fecal.
La característica dominante de este trastorno es la fotosensibilidad, quizá debida al aumento en los niveles plasmáticos de protoporfirina. El exceso de protoporfirina plasmática es causado por la sobreproducción de ésta por los hepatocitos y eritrocitos. Aunque la protoporfirina de los eritrocitos también aumenta en la anemia ferropriva y en la intoxicación por plomo, en estas dos situaciones se fija a la hemoglobina con más avidez y no se difunde al plasma ni a los tejidos, lo que explica porqué no ocurre fotosensibilidad.
En comparación con las porfirias hepáticas descritas antes, la edad de inicio de la protoporfiria eritropoyética suele ser antes de los cuatro años de edad, y el trastorno predomina en hombres. Los síntomas son precipitados por la exposición a la luz solar y se pueden limitar a sensación de quemadura y prurito, a menudo acompañados de edema, eritema o urticaria. Con menos frecuencia aparecen ampollas y ulceración; también se pueden encontrar escoriaciones secundarias al rascado. La recurrencia de estas lesiones por exposición crónica a la luz solar produce cicatrización, pigmentación alterada, liquenificación y envejecimiento prematuro de la piel.
En el hígado se depositan cantidades cada vez mayores de protoporfirinas. Aunque se ha informado de insuficiencia hepática progresiva con desarrollo de cirrosis y hepatoesplenomegalia, así como defunciones por insuficiencia hepática, la alteración leve en las pruebas de funcionamiento hepático es lo común. La descompensación hepática disminuye en forma progresiva la excreción de protoporfirina, empeorando las lesiones tanto cutáneas como hepáticas. Se han encontrado cálculos vesiculares que contienen protoporfirina en algunos pacientes y anemia hipocrómica, microcítica leve en casi la mitad. También puede existir cierta hemólisis. Ocurre una forma más grave de protoporfiria si ambos padres son heterocigotos y los hijos heredan dos genes defectuosos. Se han identificado 22 mutaciones y la mayoría de los portadores son asintomáticos.28
El diagnóstico se sospecha por la historia clínica y familiar, y se confirma por el aumento en los niveles de protoporfirina fecal y en los eritrocitos. También se encuentra fluorescencia de eritrocitos y normoblastos cuando se exponen a la banda de Soret.
Es conveniente, aunque difícil de lograr, el evitar la exposición a la luz solar para prevenir la cicatrización irreversible. La ingestión de ß-carotenos, con el objeto de alcanzar niveles sanguíneos de 500 µg/dl, disminuye la fotosensibilidad. Los efectos del tratamiento pueden tardar meses. Debido a que la síntesis de protoporfirina hepática puede también contribuir al cuadro clínico, se recomienda una dieta con alto contenido de carbohidratos, aun cuando no aparezcan en este padecimiento los ataques neuropáticos agudos característicos de la PAI.
En un paciente el tratamiento con hierro oral redujo los niveles de protoporfirina en heces y eritrocitos.29 Ocurrió resolución de la anemia y mejoría en las pruebas de función hepática a los 4 meses. Se ha postulado la producción del hem aumentó en forma no enzimática o por acción de la ferroquelatasa residual. A su vez, la formación del hem pudo haber producido reducción de la concentración de protoporfirina a un nivel menos tóxico al reprimir a la AAL sintetasa y reducir la síntesis de porfirina.29
Se ha usado el trasplante hepático para la cirrosis inducida por protoporfirina con algunos buenos resultados pero con morbilidad elevada, incluyendo quemaduras por las luces del quirófano y recurrencia de la enfermedad hepática protoporfírica.28,30
Información adicional
Puede obtenerse información adicional respecto a los recursos médicos y de laboratorio, así como información de orientación a los pacientes en la página de la American Porphyria Foundation http://www.enterprise.net/apf. La American Porphyria Foundation cobra una cantidad por entrar a su dirección.
Figura 1 Dana Burns-Pizer.
Figura 2 Marcia Kammerer
Tablas 2 y 3 Preparadas para SCIENTIFIC AMERICAN Medicine por Dr. Mark G. Pelroth y M.S. Robert Hagberg.
Bibliografía
-
Moore MR: Biochemistry of porphyria. Int J Biochem 25:1353,
1993
-
Puy H, Deybach JC, Lamoril J, et al: Molecular epidemiology
and diagnosis of PBG deaminase gene defects in acute intermittent porphyria.
Am J Hum Genet 60:1373, 1997
-
McDonagh AF, Bissell DM: Porphyria and porphyrinology—the
past fifteen years. Semin Liver Dis 18:3, 1998
-
Grandchamp B: Acute intermittent porphyria. Semin Liver Dis
18:17, 1998
-
Chretien S, Dubart A, Beaupain D, et al: Alternative transcription
and splicing of the human porphobilinogen deaminase gene result either
in tissue-specific or in housekeeping expression. Proc Natl Acad Sci U
S A 85:6, 1988
-
Mustajoki P, Kauppinen R, Lannfelt L, et al: Frequency of
low erythrocyte porphobilinogen deaminase activity in Finland. J Intern
Med 231:389, 1992
-
Tatum WO IV, Zachariah SB: Gabapentin treatment of seizures
in acute intermittent porphyria. Neurology 45:1216, 1995
-
Tenhunen R, Mustajoki P: Acute porphyria: treatment with
heme. Semin Liver Dis 18:53, 1998
-
Mustajoki P, Nordmann Y: Early administration of heme arginate
for acute porphyric attacks. Arch Intern Med 153:2004, 1993
-
Kirsch RE, Meissner PN, Hift RJ: Variegate porphyria. Semin
Liver Dis 18:33, 1998
-
Martasek P: Hereditary coproporphyria. Semin Liver Dis 18:25,
1998
-
Grandchamp B, Lamoril J, Puy H: Molecular abnormalities of
coproporphyrinogen oxidase in patients with hereditary coproporphyria.
J Bioenerg Biomembr 27:215, 1995
-
Sassa S: ALAD porphyria. Semin Liver Dis 18:95, 1998
-
McColl KEL, Thompson GG, Moore MR, et al: Chester porphyria:
biochemical studies of a new form of acute porphyria. Lancet 2:796, 1985
-
Norton B, Lanyon WG, Moore MR, et al: Evidence for involvement
of a second genetic locus on chromosome 11q in porphyrin metabolism. Hum
Genet 91:576, 1993
-
Doss MO: New form of dual porphyria: coexistent acute intermittent
porphyria and porphyria cutanea tarda. Eur J Clin Invest 19:20, 1989
-
Sturrock ED, Meissner PN, Maeder DL, et al: Uroporphyrinogen
decarboxylase and protoporphyrinogen oxidase in dual porphyria. S Afr Med
J 76:405, 1989
-
Conry-Cantilena C, Vilamidou L, Melpolder JC, et al: Porphyria
cutanea tarda in hepatitis C virus–infected blood donors. J Am Acad Dermatol
32:512, 1995
-
Elder GH: Hepatic porphyrias in children. J Inher Metab Dis
20:237, 1997
-
Roberts AG, Whatley SD, Morgan RR, et al: Increased frequency
of the haemochromatosis Cys282Tyr mutation in sporadic porphyria cutanea
tarda. Lancet 349:321, 1997
-
Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, et al: Porphyria
cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology
27:1661, 1998
-
Gibertini P, Rocchi E, Cassanelli A, et al: Advances in the
treatment of porphyria cutanea tarda: effectiveness of slow subcutaneous
desferrioxamine infusion. Liver 4:280, 1984
-
Sheikh MY, Wright RA, Burruss JB: Dramatic resolution of
skin lesions associated with porphyria cutanea tarda after interferon-alpha
therapy in a case of chronic hepatitis C. Dig Dis Sci 43:529, 1998
-
Desnick RJ, Glass IA, Xu W, et al: Molecular genetics of
congenital erythropoietic porphyria. Semin Liver Dis 18:77, 1998
-
Pimstone NR, Gandhi SN, Mukerji SK: Therapeutic efficacy
of oral charcoal in congenital erythropoietic porphyria. N Engl J Med 316:390,
1987
-
Mazurier F, Moreau-Gaudry F, Salesse S, et al: Gene transfer
of the uroporphyrinogen III synthase cDNA into haematopoietic progenitor
cells in view of a future gene therapy in congenital erythropoietic porphyria.
J Inherit Metab Dis 20:247, 1997
-
Taketani S, Fujita H: The ferrochelatase gene structure and
molecular defects associated with erythropoietic protoporphyria. J Bioenerg
Biomembr 27:231, 1995
-
Cox TM, Alexander GJ, Sarkany RP: Protoporphyria. Semin Liver
Dis 18:85, 1998
-
Gordeuk VR, Brittenham GM, Hawkins CW, et al: Iron therapy
for hepatic dysfunction in erythropoietic protoporphyria. Ann Intern Med
105:27, 1986
-
Bloomer JR, Rank JM, Payne WD, et al: Follow-up after liver
transplantation for protoporphyric liver disease. Liver Transpl Surg 2:269,
1996