Neurología
⭳ Abrir artículo (PDF)1.1 MBEste artículo es idéntico en la Edición 3/2000.
Contenido del artículo
XV ENFERMEDAD DE PARKINSON Y TRASTORNOS DE MOVIMIENTO RELACIONADOS CON LOS GANGLIOS BASALES
- Fisiopatología de los trastornos del movimiento
- Trastornos hipoquinéticos
- ENFERMEDAD DE PARKINSON IDIOPATICA
- Etiología
- Patogenia
- Características clínicas
- Condiciones asociadas
- Tratamiento
- Tratamiento de la enfermedad de Parkinson avanzada
- OTRAS FORMAS DE PARKINSONISMO
- Trastornos hiperquinéticos
- Temblor fisiológico y esencial
- Mioclonías
- Trastornos del movimiento inducido por medicamentos
XV ENFERMEDAD DE PARKINSON Y TRASTORNOS DE MOVIMIENTO RELACIONADOS CON LOS GANGLIOS BASALES
DR. JORGE L. JUNCOS
DR. MAHLON R. DELONG
La mayoría de los trastornos de movimiento se deben a enfermedades que afectan los ganglios basales. Los trastornos hipoquinéticos se caracterizan por alteración de los movimientos voluntarios, el parkinsonismo es un ejemplo. Los trastornos hiperquinéticos se presentan como diversos síndromes clínicos, incluyendo corea, balismo, distonía, temblor, tics y mioclonías.
Cada trastorno de movimiento típicamente tiene múltiples etiologías pero fisiopatología y fenotipo compartido. En ausencia de marcadores biológicos y patrones de herencia claros, el diagnóstico etiológico de la mayoría de los trastornos de movimiento es un ejercicio clínico muy difícil. Sin embargo, los adelantos importantes en la comprensión de la organización funcional de los ganglios basales y en la fisiopatología de los trastornos del movimiento ha originado el desarrollo de nuevos enfoques farmacológicos y quirúrgicos para el tratamiento.
Los trastornos de movimiento rara vez son trastornos solo del movimiento. Las alteraciones psiquiátricas y cognitivas asociadas pueden contribuir en forma significativa e incluso dominar en el cuadro clínico. Es importante detectar la morbilidad psiquiátrica concomitante porque ésta puede contribuir en forma desproporcionada al trastorno. Por ejemplo, la aparición de demencia y depresión puede causar deterioro clínico inexplicable a pesar del tratamiento adecuado de los síntomas motores.
Fisiopatología de los trastornos del movimiento
Los trastornos de movimiento que se originan en los ganglios basales parecen depender de disfunción del circuito motor ganglios basales-tálamocortical [ver figura 1].1 Las alteraciones de las señales de salida de los ganglios basales a nivel del segmento interno del globo pálido (GPi) puede causar trastorno en los movimientos tanto voluntarios como involuntarios.23
Se ha desarrollado un modelo fisiopatológico de parkinsonismo con base en estudios animales [ver figura 2]. La característica bioquímica de este síndrome es la deficiencia de dopamina, que promueve una actividad excesiva de la llamada vía indirecta. Esto aumenta el estímulo excitatorio sobre el GPi desde el núcleo subtalámico.2 La mayor influencia inhibitoria del GPi ocasiona mayor inhibición de las neuronas tálamocorticales, lo que provoca que algunas áreas de proyección cortical tengan menos respuesta a estímulos que en condiciones normales participan en el inicio y ejecución del movimiento. Por ejemplo, la aquinesia y la bradicinesia son causadas por inhibición excesiva de las neuronas tálamocorticales. El apoyo para este modelo deriva de estudios que demuestran que la inactivación selectiva del núcleo subtalámico o GPi reduce mucho los signos motores de la enfermedad de Parkinson (EP), incluyendo la aquinesia.4,5 Además, se ha demostrado el restablecimiento en pacientes de la activación cortical durante una tarea de movimiento por medio de mediciones en tomografía de emisión de positrones (TEP) antes y después de la palidotomía, un procedimiento que destruye quirúrgicamente el segmento motor del GPi.6
En contraste con los trastornos hipoquinéticos, los hiperquinéticos son resultado de un estímulo disminuido del GPi [ver figura 2].7 En este caso las neuronas tálamocorticales, que no tienen el impulso tónico inhibidor normal proveniente del GPi, se vuelven más sensibles a los estímulos corticales o muestran una mayor tendencia a descargar en forma espontánea, lo que ocasiona movimientos deteriorados o involuntarios. Tanto en el hemibalismo, que es causado de lesiones del núcleo subtalámico, como en la enfermedad de Huntington (EH), en la que ocurre una pérdida selectiva temprana de las neuronas estriadas de la vía indirecta, la menor salida de estímulos del núcleo subtalámico causa actividad anormalmente baja del GPi y menores niveles de inhibición talámica.8
Existen evidencias contundentes acerca de que los trastornos hipo o hiperquinéticos que clínicamente son contrastantes, son causados en forma respectiva por niveles excesivos o reducidos de estímulos del GPi del circuito motor, con inhibición o desinhibición talámica que ocasiona los síndromes respectivos. Cambios fisiopatológicos semejantes son responsables de los trastornos de los movimientos oculares que ocurren después del daño al circuito oculomotor tanto en la EP como en la EH. Es tentador especular sobre si ocurren cambios patológicos comparables en otros circuitos ganglios basales-tálamo para producir los complejos trastornos de comportamiento, cognitivos y límbicos equivalentes de los síntomas y signos motores que ocurren en la disfunción del circuito motor (v.gr., los síndromes del lóbulo frontal, el pensamiento lento [bradifrenia], la manía y la depresión, los tics y los trastornos obsesivo-compulsivos).9
Trastornos hipoquinéticos
El parkinsonismo es un síndrome clínico con múltiples etiologías que se caracteriza por grados variables de discinesia (movimiento lento), temblor, rigidez y inestabilidad postural.10 El común denominador de todas las formas de parkinsonismo es la reducción de la trasmisión dopaminérgica estriada como resultado de disrupción estructural o funcional de las vías nigroestriadas. El parkinsonismo puede dividirse en enfermedad de Parkinson idiopática, que es responsable de alrededor del 75 porciento de los casos, y formas secundarias de parkinsonismo [ver tabla 1]. Es importante distinguir la EP de otras formas de parkinsonismo porque el pronóstico y tratamiento son diferentes. El diagnóstico temprano y adecuado se vuelve incluso más importante cuando existen tratamientos que pueden retrasar o detener la progresión del proceso patológico subyacente.
ENFERMEDAD DE PARKINSON IDIOPATICA
Etiología
La enfermedad de Parkinson es un trastorno neurodegenerativo de etiología incierta que afecta a más de un millón de personas en los Estados Unidos, alrededor de uno porciento de los mayores de 55 años.11 Es responsable de casi tres cuartas partes de los casos de parkinsonismo. Los papeles relativos de los factores hereditario, ambiental y endógeno son inciertos. Varios estudios epidemiológicos y familiares recientes indican que la historia familiar positiva, cuando se investiga en forma adecuada y detallada, es mucho más común de lo que se pensaba antes. En parte con base en la revisión de datos obtenidos de estudios en gemelos monocigotos, en la actualidad parece ser que los pacientes con EP pueden heredar una vulnerabilidad genética para la enfermedad.12 Sin embargo, la manifestación del fenotipo depende del proceso de envejecimiento (v.gr., pérdida relacionada con la edad de la capacidad de fosforilación oxidativa) y de una serie de otros factores de desarrollo, ambientales y genéticos no identificados. Los estudios epidemiológicos han demostrado que el riesgo de EP es mayor entre quienes viven en áreas rurales, beben agua de pozo o están expuestos a pesticidas.13 El consenso actual es que es muy probable que la EP sea causada por una interacción entre factores genéticos, ambientales y estrés oxidante endógeno.
En un artículo clave, una mutación en el gen alfa-sinucleína del cromosoma 4 se asoció con una forma autosómico dominante de EP en un pedigree italiano extenso y en tres familias no relacionadas de origen griego.14 La mutación consiste en la sustitución de una base en la posición 209 (G209T). El gen codifica la proteína precursora del componente amiloide no § de las placas neuríticas de la enfermedad de Alzheimer. La proteína alfa-sinucleína se encuentra en las terminales presinápticas y su distribución en el cerebro es semejante a la de los cuerpos de Lewy (la presencia de inclusiones citoplasmáticas prominentes).14 Se ha postulado que la proteína mutada puede tener una estructura terciaria anormal, lo que la hace propensa a autoagregarse y a formar núcleos semejantes a amiloide, como los encontrados en los cuerpos de Lewy y en las placas dendríticas.14 Aunque la mayoría de los casos no son autosómico dominantes, este descubrimiento puede orillar a un nuevo conocimiento fundamental sobre la fisiopatología de la muerte de las células dopaminérgicas, que ocurre en todos los pacientes con EP.
Patogenia
Las características patológicas de la EP son la degeneración de las células dopaminérgicas de la sustancia negra compacta (SNc) y de los cuerpos de Lewy en las neuronas pigmentadas del tallo cerebral, incluyendo el locus ceruleus, el núcleo del rafé pontino el núcleo motor dorsal del nervio vago.15 Aunque la depleción celular en la SNc y otras áreas ocurre en forma gradual durante muchos años, los síntomas se desarrollan solo cuando se han perdido entre el 50 y el 80 porciento de las células y han fallado los mecanismos compensatorios.
Se ha puesto gran atención al papel del estrés oxidativo en la EP y al daño que pueden causar los radicales libres producidos por el metabolismo de la dopamina y otras sustancias.16 La disfunción mitocondrial es otro factor posible que puede contribuir a la vulnerabilidad al estrés oxidativo, y se ha reportado evidencia de disfunción mitocondrial en el músculo y las plaquetas de los pacientes con EP.17 En la actualidad, el papel de la disfunción mitocondrial en la EP es un punto de gran interés y controversia.
Características clínicas
Lo más común es que la EP se desarrolle en forma insidiosa hacia la mitad de la vida, con un pico en la edad de inicio entre los 55 y los 65 años [ver figura 3]. En los estadios iniciales de la enfermedad el diagnóstico puede ser difícil, a menos que exista el temblor en reposo característico de la enfermedad. En ausencia de temblor, el diagnóstico puede pasar desapercibido si la atención se enfoca en lo que parece debilidad, molestias musculoesqueléticas inespecíficas o problemas neuromusculares periféricos. Las características clínicas de la EP son bradicinesia, rigidez muscular y un temblor en reposo de 4 a 5 Hz. La presencia de dos de estos tres signos cardinales y una respuesta clínica clara a la levodopa, el precursor de la dopamina, son suficientes para hacer el diagnóstico de probable EP.18 El inicio unilateral de los síntomas y la presencia de temblor en reposo favorece en forma intensa el diagnóstico de EP. La fascies inexpresiva, el menor parpadeo, la postura inclinada, los movimientos lentos y la micrografia (escritura que se deteriora y disminuye de tamaño) suelen ser frecuentes en las fases tempranas de la enfermedad.
La bradicinesia, o retraso en la ejecución de movimientos, y los movimientos escasos (acinesia) son de las características más incapacitantes de la EP porque interfieren con todos los aspectos de la vida diaria, incluyendo actividades como caminar, levantarse de una silla, voltearse en la cama o vestirse. También se afecta el control fino, que se manifiesta por menor destreza de los dedos y micrografía. La hipofonía y el lenguaje monotónico son también manifestaciones de bradicinesia.
El temblor en reposo comienza típicamente en la porción distal del brazo, en donde se presenta como un movimiento constante de «frota monedasÕ. El temblor suele progresar en dirección proximal en el brazo y después en la pierna antes de cruzar hacia el otro lado, lo que ocurre después de un año o más. Puede afectar también la cara, lengua y mandíbula, pero típicamente el Parkinson no afecta solo la cabeza, como el temblor esencial. A diferencia de los temblores esenciales y cerebelosos, el temblor parkinsónico suele presentarse en reposo y atenuarse o desaparecer con el movimiento. Puede reaparecer al terminar el movimiento. El temblor postural puede preceder y coexistir con el temblor en reposo, y ambos son comunes con el envejecimiento.
La rigidez representa un aumento uniforme en la resistencia a los movimientos pasivos durante todo el rango de movimiento de los grupos musculares que actúan sobre una articulación. Puede haber interrupciones breves y regulares de la resistencia durante el movimiento pasivo, que corresponden a temblor subclínico y pueden dar la sensación de una rueda dentada. La combinación de rigidez y distonía típicamente provoca flexión del cuello, inclinación hacia adelante de la porción superior del tronco y tendencia a mantener los brazos en una posición en flexión y aducción.
La inestabilidad postural es una de las características más limitantes y refractarias del parkinsonismo y es un factor contribuyente importante para las caídas, lesiones y menor locomoción. Aunque puede observarse disminución en los reflejos posturales al principio de la EP, rara vez contribuye un problema hasta que la enfermedad progresa. De hecho, la inestabilidad postural significativa en fases tempranas del padecimiento sugiere un diagnóstico diferente a EP.
La marcha parkinsónica se caracteriza por pasos cortos, arrastrados y con tendencia a voltearse en bloque. En las fases avanzadas de la enfermedad se dificulta el iniciar, detener la marcha y voltearse. Puede ocurrir marcha festinante, en un esfuerzo aparente del paciente por alcanzar el centro de gravedad de su cuerpo. Sin embargo, la presencia de una marcha atáxica o atípica sugiere otro diagnóstico, como hidrocefalia o atrofia sistémica múltiple con afección cerebelosa. El quedarse inmóvil "congelado" es una característica de la EP avanzada y ocurre sobre todo en relación con el inicio de la marcha (duda para iniciar), al intentar cambiar de dirección o al entrar en un área muy concurrida o angosta. A diferencia de la mayoría de los signos motores parkinsónicos, esta alteración puede exacerbarse con el tratamiento dopaminomimético en dosis altas.
Condiciones asociadas
Además del parkinsonismo, muchos pacientes presentan alteraciones cognitivas, de ánimo, sensoriales, de sueño y autonómicas. Los cambios en la función cognitiva, de comportamiento y de ánimo son resultado en parte de la alteración de otros circuitos no motores de los ganglios basales. Aunque la demencia no es una característica temprana de la EP, ocurre hasta en el 30 porciento de los pacientes.19,20 La demencia parece ocurrir con más frecuencia en pacientes que tienen características principalmente aquinéticas-rígidas y suele ser precedida por alucinaciones inducidas por medicamentos y depresión. Desde el punto de vista patológico, los pacientes con demencia tienen patologías mixtas, que pueden incluir enfermedad de Alzheimer y cambios de enfermedad difusa de los cuerpos de Lewy.
La depresión afecta hasta al 50 porciento de los pacientes con EP. Son comunes los cambios de personalidad, caracterizados por apatía, ausencia de asertividad e indecisión. Puede ocurrir depresión en cualquier fase de la enfermedad, y correlaciona poco con la severidad de la misma.21-23 Debido a que el retraso psicomotor de la depresión puede ser muy semejante a las características hipoquinéticas del parkinsonismo, la depresión puede ser difícil de detectar. Esta puede ser inducida o agravada en forma iatrogénica por la polifarmacia antiparkinsónica. Por lo tanto, debe consderarse la simplificación del tratamiento antes de que el médico decida agregar farmacoterapia para la depresión.
Los trastornos del sueño son comunes en la EP. Muchos pacientes no duermen suficiente y otros tienen sueño de mala calidad. El sueño de movimientos oculares rápidos suele estar muy fragmentado. La somnolencia diurna y las siestas frecuentes son signos típicos de interrupción del sueño. Los factores que alteran el sueño incluyen reaparición del parkinsonismo durante la noche en forma de bradicinesia y rigidez (se dificulta el voltearse en la cama) y temblor o movimientos involuntarios (v.gr., mioclonías y movimientos periódicos de las piernas) que se deben a la reducción del efecto de los medicamentos antiparkinsónicos.24 Los sueños vívidos y las alucinaciones, que pueden ser efectos adversos del tratamiento antiparkinsónico, pueden contribuir también a los trastornos del sueño.25 Además de las alteraciones relacionadas con la enfermedad y los medicamentos, con frecuencia ocurren apnea del sueño y otros trastornos en los pacientes ancianos.24 Los datos sobre las características del sueño suelen brindar información útil, y el tratamiento exitoso de estos trastornos mejora en forma típica la capacidad funcional durante el día. Otras alteraciones no motoras en la EP incluyen disfunción sexual, disfunción urinaria y constipación.
Tratamiento
Los objetivos del tratamiento son mantener la función controlando la alteración primaria con tratamiento sintomático y minimizar la incapacidad secundaria (contracturas, artritis acelerada) por medio de un programa constante de ejercicio físico. Ocurrió un gran revuelo en 1969, cuando Cotzias demostró que la administración oral del precursor de dopamina levodopa podía aliviar el parkinsonismo.26 El tratamiento con levodopa se convirtió en la base del tratamiento y mejoró mucho la calidad de vida de los pacientes con EP, proporcionándoles una expectativa de vida casi normal. Sin embargo, pronto fue evidente que la levodopa no detenía la progresión de la enfermedad y, más alarmante, que el beneficio terapéutico se asociaba con complicaciones motoras y cognitivas asociadas con el fármaco.27,28
Se cree que las estrategias terapéuticas tempranas afectan en forma significativa la progresión tardía de los síntomas y que el manejo equivocado de estas estrategias puede contribuir a inducir incapacidad secundaria. Sin embargo, existe controversia sobre qué constituye el manejo farmacológico temprano óptimo de la EP.29 Los puntos de preocupación son la contribución potencial de la dopamina para acelerar la progresión de la enfermedad a través de la formación de radicales libres tóxicos y el desarrollo tardío de complicaciones motoras relacionadas con los fármacos.30,31 Sin embargo, existen pocas evidencias clínicas convincentes que apoyen estas preocupaciones. Para evaluar estos aspectos un estudio patrocinado por los Institutos Nacionales de Salud (NIH de los EUA) investiga el uso de dosis altas y bajas de carbidopa-levodopa.
Desde un punto de vista práctico, la levodopa debe iniciarse en cuanto el paciente encuentre que los síntomas interfieren con su empleo o estilo de vida. Las estrategias para ahorrar levodopa, como retrasar su inicio e iniciar agonistas de dopamina u otros agentes menos potentes en las fases tempranas de la enfermedad, pueden estar justificadas en algunos casos. Sin embargo, en general, la monoterapia con estos agentes rara vez tiene el mismo valor terapéutico que el tratamiento con levodopa. Esta opinión puede requerir reevaluación al introducirse los nuevos agonistas de dopamina de acción prolongada que se supone controlan en forma adecuada los síntomas y los nuevos monoamino oxidasa tipo B (MAO B) e inhibidores de la MAO AB.
El tratamiento sintomático de la EP debe individualizarse. Por ejemplo, los pacientes con inicio temprano representan una categoría especial. En ellos es importante descartar el parkinsonismo secundario, como la enfermedad de Wilson, la ataxia espinocerebelosa tipo 3 (enfermedad de Machado-Joseph) o la EP inducida por medicamentos [ver tabla 1]. Los autores recomiendan que estos pacientes sean referidos a un especialista en trastornos del movimiento para evaluación y tratamiento. Es importante tratar de retrasar el desarrollo de complicaciones motoras inducidas por la levodopa en los pacientes jóvenes.
La carbidopa-levodopa es la mejor elección para el tratamiento inicial de todos los pacientes con EP. La carbidopa es un inhibidor periférico decarboxilasa que bloquea la ruptura periférica de la levodopa. Por lo tanto, la carbidopa minimiza los efectos adversos asociados con la conversión periférica de levodopa a dopamina. Aunque no existen evidencias para elegir un esquema diferente a la carbidopa-levodopa en dosis bajas, el tratamiento combinado temprano (v.gr., con amantadina, anticolinérgicos o agonistas dopaminérgicos) puede estar justificado mientras controle los síntomas en forma adecuada.
Levodopa y sus fórmulas La levodopa está disponible en fórmulas de liberación inmediata regular de carbidopa más levodopa (10 mg de carbidopa/25 mg de levodopa, 25 mg/100 mg y 25 mg/250 mg) y de liberación controlada (LC) (25 mg/100 mg, 50 mg/200 mg). Los suplementos de carbidopa (25 mg al día a 200 mg al día) se usan en ocasiones para tratar a algunos pacientes con náusea persistente, al parecer en respuesta a dosis inadecuadas de carbidopa. Para pacientes con náusea persistente el antiemético débil trimetobenzamida (para pacientes externos) y el ondansetrón intravenoso u oral (para hospitalizados) son bien tolerados y eficaces. Otros antieméticos, como los antagonistas del receptor de dopamina, proclorperacina y metoclopramida, deben evitarse porque pueden empeorar en forma importante los síntomas parkinsónicos.
El tratamiento con carbidopa-levodopa de LC permite la comodidad de menos dosis durante el día y una respuesta clínica más gradual en pacientes con fluctuaciones motoras leves a moderadas (supresión).32 En un estudio doble ciego de cinco años de duración que comparó la fórmula carbidopa-levodopa de liberación inmediata regular con la controlada, la segunda fue más eficaz para disminuir la progresión de la incapacidad, medida por varios indicadores de calidad de vida. Sin embargo, sorprendentemente, pareció que la carbidopa-levodopa LC no tuvo ventajas sobre la regular para disminuir la incidencia de fluctuaciones motoras.33 Solo el 16 porciento de los pacientes en ambos grupos desarrollaron fluctuaciones motoras, un porcentaje muy bajo comparado con reportes más tempranos. Esta baja incidencia de discinesias puede deberse en parte con las dosis relativamente bajas de levodopa que se han usado (200 a 400 mg/día).
El inicio del tratamiento con las fórmulas de uso regular o de LC de carbidopa-levodopa es aceptable. El precio de la forma regular, que en la actualidad está disponible en su fórmula genérica, es menor que el de la fórmula de LC. Aunque la carbidopa-levodopa regular se absorbe mejor con el estómago vacío, suele administrarse con los alimentos para disminuir la náusea. Por el contrario, la absorción y biodisponibilidad de las fórmulas de LC aumenta cuando se toma con alimentos.32 Los efectos adversos de la carbidopa-levodpa pueden controlarse iniciando con una dosis pequeña (v.gr., media tableta de carbidopa-levodopa, 25 mg/100 mg bid) y después aumentando la dosis según se tolere hasta que se presente náusea. El ajuste de la dosis debe hacerse despacio porque la respuesta total a cada incremento se presenta en varias semanas. En la mayoría de los casos la dosis de levodopa no debe exceder 300 a 400 mg/día en las fases tempranas de la enfermedad. Pueden requerirse dosis más altas y más frecuentes en pacientes con enfermedad más avanzada que presentan fluctuaciones motoras.
Si se selecciona la carbidopa-levodopa LC como tratamiento inicial, la dosis es la mitad de una tableta de 50 mg/200 mg dos veces al día. Los autores prefieren que el paciente inicie tomando el medicamento en el desayuno y la comida, con un intervalo de cinco a seis horas en lugar del esquema estricto de nueve a 12 horas. El objetivo es obtener una meseta en la concentración plasmática de levodopa en la parte inicial del día y dejar que esta meseta se reduzca durante el resto del día. Eventualmente el paciente requerirá dosis en la tarde y noche. Debido a la menor biodisponibilidad de la levodopa en la fórmula de LC (77 porciento en comparación con la fórmula regular), la fórmula de LC de 600 mg equivale a alrededor de 400 mg de la fórmula de liberación inmediata. Por último, se ha reportado que la fórmula de LC induce o agrava los síntomas distónicos, comparado con la carbidopa-levodopa regular, en especial en pacientes con inicio temprano.32
Estrategias para aumentar la levodopa Otras estrategias para aumentar la trasmisión de dopamina incluyen el bloqueo metabólico de la dopamina por inhibición de la enzima MAO B.34 La seliginina es un inhibidor selectivo y reversible de la MAO B que tiene un efecto sintomático débil cuando se usa solo y un efecto sintomático modesto cuando se usa con carbidopa-levodopa. La seliginina se emplea para aliviar la supresión leve o tumor residual y evitar aumentar la dosis de levodopa. Está disponible en cápsulas o tabletas de 5 mg. La dosis habitual es de 5 mg con el desayuno y la comida. Mientras se mantenga esta dosis, no se requieren restricciones dietéticas paa evitar las crisis hipertensivas inducidas por tiramina, a diferencia de lo que sucede con los inhibidores no selectivos (MAO AB) y los inhibidores de la MAO A, para los que se requiere restricción dietética. El insomnio es un efecto adverso significativo de la seliginina, en especial cuando se toma después de mediodía. Las personas mayores y las que tienen enfermedad cardiaca estable pero importante pueden beneficiarse de dosis tan bajas como 2.5 mg/día. La dosis debe disminuirse o eliminarse si ocurren efectos adversos hiperdopaminérgicos refractarios (v.gr., empeoramiento de la discinesia, alucinosis o confusión).
Una estrategia novedosa puede aumentar la duración de la respuesta a la levodopa y aliviar las fluctuaciones motoras.35 La O-metilación de la levodopa a 3-O-metildopa por la catecol-O-metiltransferasa (COMT) hace que la levodopa esté menos disponible para conversión a dopamina y por tanto sea menos eficaz. Debido a que se encuentra actividad de COMT en el tejido periférico y el cerebro, los inhibidores de la COMT aumentan la absorción de la levodopa y disminuyen su eliminación. El retraso en la eliminación de la levodopa puede reducir las fluctuaciones motoras clínicas y los requerimientos de levodopa. El entacapone y el tolcapone son dos inhibidores de la COMT que se evalúan en la actualidad en estudios de fase III.
Agonistas de dopamina Los agonistas de dopamina no requieren transformación o transporte facilitado a través de la barrera hematoencefálica, y actúan directamente sobre los receptores posinápticos de dopamina. Dos de éstos, la bromocriptina y el pergolide, actúan mejor como adyuvantes del tratamiento con levodopa y no son tan eficaces como ésta última. Sus efectos adversos (confusión, alucinaciones, trastornos del sueño) y la relación riesgo-beneficio son menos favorables que los de la carbidopa-levodopa. Cuando los agonistas de dopamina se usan como monoterapia en tratamientos ahorradores de levodopa, la mayoría de los pacientes requieren de la adición de levodopa después de seis a 18 meses para mantener un control adecuado de los síntomas. Además, un estudio extenso no pudo demostrar ningún beneficio a largo plazo de la monoterapia temprana con agonistas.36 Dos agonistas nuevos, el pramipexol y el ropinirol, pueden ser mejores agentes para monoterapia y tener acción más prolongada. La dosis inicial del pramipexol es de 0.125 mg tres veces al día. Al final de la primera semana la dosis puede aumentarse a 0.25 mg tres veces al día, con incrementos semanales de 0.25 mg tres veces al día, hasta una dosis máxima de 1.5 mg tres veces al día, con o sin levodopa. La dosis inicial del ropinirol es de 0.25 mg tres veces al día. Dependiendo de la respuesta, la dosis puede aumentarse en forma semanal 0.25 mg tres veces al día hasta 1 mg tres veces al día. Después de la semana 4 la dosis diaria puede aumentarse a 1.5 mg/día en forma semanal, hasta una dosis máxima de 9 mg/día, y después aumentarse 3 mg/día hasta una dosis máxima de 24 mg/día. El uso de los agonistas de dopamina en las fases tardías de la enfermedad se justifica en casos seleccionados porque estos agentes pueden tener fluctuaciones motoras suaves y pueden disminuir las discinesias. Sin embargo, esto requiere de un ajuste cuidadoso de la dosis y de ajustes simultáneos en el resto del tratamiento antiparkinsónico.
Amantandina y anticolinérgicos Algunos médicos prefieren tratar la EP leve con agentes menos potentes, como la amantadina y anticolinérgicos, para retrasar el inicio de la levodopa, incluso a pesar de que no existen datos de que esta maniobra evite la aparición de las discinesias. Sin embargo, es lógico iniciar el tratamiento con estos agentes menos potentes en los pacientes jóvenes con EP, en especial en los que tienen distonía importante. Los pacientes con temblor predominante pueden también responder en forma satisfactoria a la amantadina y a los anticolinérgicos.
El efecto protector potencial de la amantadina puede ser mediado por su débil efecto antagonista del glutamato. En la actualidad se evalúan antagonistas del glutamato más potentes y selectivos para determinar su eficacia como adyuvantes en el tratamiento antiparkinsónico.37 Un estudio clinicopatológico reciente sugiere que la amantadina puede tener también un efecto neuroprotector sobre la progresión de los síntomas y la muerte de las células del área negra.38 Por último, se ha sugerido que la amantadina es un adyuvante útil a la levodopa en fases más avanzadas de EP y puede reducir la severidad de las discinesias inducidas por medicamentos. Los efectos adversos de la amantadina son edema, eritema y livedo reticularis. En los pacientes ancianos puede agravar la confusión y la psicosis. La dosis inicial es de una tableta de 100 mg dos veces al día; en caso necesario la dosis puede aumentarse a tres veces al día después de una semana. Debido a que la amantadina se elimina por los riñones, puede acumularse con rapidez hasta niveles tóxicos en pacientes con daño renal.
Los anticolinérgicos se han usado por mucho tiempo en la EP, en especial por sus efectos sobre el temblor, la rigidez y la distonía. Sus efectos sobre la acinesia, la bradicinesia, la postura y la marcha son limitados. Las principales desventajas, en especial en los ancianos, son el deterioro de la memoria, las alucinaciones, la visión borrosa, la constipación, la polaquiuria y la xerostomía. Este último efecto adverso puede ser útil en los pacientes con sialorrea importante, que se asocia con frecuencia con la EP.
Tratamiento neuroprotector Los mecanismos de la muerte de las células del área negra parecen incluir el estrés por oxidantes, que suele agravarse por el alto contenido de los compuestos reactivos, dopamina y melanina, en estas células. La selegilina tiene propiedades antioxidantes in vitro. En estudios experimentales en animales, la seligilina puede bloquear la neurotoxicidad dopaminérgica de la 1-metil-4-fenil-1,2,3,6-tetrahidropiridina (MPTP) al bloquear en forma selectiva la formación del metabolito de la MPTP, el ión 1-metil-4-fenilpiridinio (MPP+), una toxina con alto poder oxidativo para las neuronas de dopamina.17,39 Debido a que el MPTP también produce un síndrome semejante a la EP en humanos, se ha postulado que la selegilina podría reducir las fuentes intrínsecas y extrínsecas de estrés oxidante en las neuronas dopaminérgicas humanas. Su uso en la EP como agente neuroprotector fue apoyado por los reportes del grupo DATATOP (Tratamiento del parkinson con tratamiento antioxidante con tocoferol y deprenil), que demostraron que la monoterapia con selegilina retrasaba en forma significativa la progresión de los síntomas durante un año.39,40 La interpretación de estos resultados se confundió con un efecto sintomático leve e inesperado de la selegilina que pudo haber causado el retraso de la progresión de los síntomas cuando se comparó con el grupo tratado con placebo.17 El seguimiento y el análisis posterior de los datos del DATATOP y de varios estudios pequeños sobre selegilina no han revelado ninguna reducción sustancial inducida por selegilina en la progresión de los síntomas después de un año.41-43 Por lo tanto, los datos clínicos disponibles no apoyan el concepto de que la selegilina pueda tener un efecto benéfico sobre la EP a largo plazo. Un solo estudio europeo reportó mayor mortalidad con la seleginina, pero este dato no concuerda con los estudios previos, por lo que es poco confiable.44
Tratamiento de la enfermedad de Parkinson avanzada
El manejo clínico de la EP avanzada se vuelve cada vez más complejo al empeorar la marcha y existir inestabilidad postural, con aumento de los periodos estáticos, las caídas y la aparición de fluctuaciones motoras y discinesias. Igualmente incapacitantes son los periodos confusionales inducidos por medicamentos, las alucinaciones y la psicosis. En esta fase está indicado referir al paciente con un especialista en trastornos del movimiento. Lo siguiente es solo una breve descripción de los aspectos más importantes en el manejo de la EP avanzada.
Supresión Típicamente, la supresión de la acción de la levodopa es el signo más temprano de fluctuaciones motoras.27 La administración de medicamentos antiparkinsónicos antes de los alimentos en lugar de con ellos puede evitar la absorción errática y parcial que contribuye a este problema. La carbidopa-levodopa regular puede combinarse con la fórmula de liberación controlada (v.gr., media tableta de liberación inmediata de carbidopa-levodopa 10/100 mg y una tableta de LC de 50/200 mg) para proporcionar un mejor balance entre las respuestas inmediata y sostenida. Los agonistas de la dopamina y la selegilina tienen una vida media más larga que la carbidopa-levodopa, y producen una respuesta clínica más suave que la primera usada en forma aislada en los pacientes que presentan fluctuaciones motoras. Otra estrategia consiste en acortar los intervalos de las dosis. Sin embargo, los esquemas con dosis más frecuentes que cada cuatro horas son difíciles de manejar desde el punto de vista clínico y probablemente deben usarse solo en clínicas especializadas de trastornos del movimiento.
Fenómeno presente-ausente Ocurren cambios súbitos e impredescibles en las respuestas antiparkinsónicas mezcladas con discinesias, descargas autonómicas (diaforesis profusa, urgencia urinaria) y cambios en el estado de ánimo en algunos pacientes con EP avanzada, a lo que se le llama fenómeno de presente-ausente (on-off).45 Las estrategias para controlar la supresión (ver antes) suelen ser de valor muy limitado en la mayoría de los pacientes que presentan este fenómeno. Se requieren estrategias más agresivas para estabilizar la variabilidad en la concentración en plasma de la dopamina y por lo tanto la respuesta clínica. Estas estrategias incluyen dietas de redistribución de proteínas y, por lo tanto, respuesta clínica. Las dietas consisten en mantener la ingesta proteína y calórica óptima diaria cambiando el consumo de proteínas principalmente a las comidas de la tarde. Esto minimiza la competencia diurna entre la levodopa y otros aminoácidos neutros grandes por el transporte a través de la barrera hematoencefálica. Los pacientes que sufren del fenómeno presente-ausente son también candidatos ideales para la neurocirugía estereotáctica.
Cambios en el comportamiento inducidos por drogas Los pacientes con EP pueden sufrir cambios en la personalidad inducidos por los medicamentos que son lentos pero profundos, y que se expresan como actitudes temperamentales, ilógicas y demandantes, egocentrismo y aparente desinterés por las necesidades de otros. Esto puede ser un preludio de una depresión agitada o psicosis inducida por los medicamentos o secundaria a la aparición de demencia. La psicosis inducida por drogas se caracteriza por alucinaciones visuales con retención del yo interno. las alucinaciones auditivas sugieren depresión psicótica coexistente o demencia, pero pueden ser un efecto adverso de los medicamentos anticolinérgicos.47 En muchos casos los síntomas cognitivos y psiquiátricos cesan con la eliminación de los anticolinérgicos y la amantadina. Sin embargo, en algunos pacientes es necesario disminuir las dosis de dopaminomiméticos o suspenderlos, típicamente en el siguiente orden: selegilina, dosis noctura de antagonista de dopamina o carbidopa-levodopa de LC y, por último, carbidopa-levodopa regular. Si el paciente mejora después de un ajuste en la polifarmacia, el impacto sobre los síntomas parkinsónicos motores irá de negligible a positivo. Si el paciente requiere una reducción drástica en el tratamiento antiparkinsónico, es probable que el empeoramiento de los síntomas motores sea intolerable. No se recomiendan ya los días aislados sin levodopa porque varios reportes indican que son potencialmente peligrosos y que no proporcionan un beneficio significativo a largo plazo.
Cuando la reducción en la polifarmacia antiparkinsónica es intolerable, puede requerirse un medicamento antipsicótico, y la clozapina es el fármaco de elección en la mayoría de los pacientes. La clozapina es un antipsicótico atípico muy eficaz, con propiedades bloqueadoras de los receptores dopaminérgico (D4) y serotoninérgico (S2) y pocos efectos adversos extrapiramidales. La clozapina puede tener efectos adicionales antitemblor y antidiscinéticos. Sus efectos adversos más persistentes son hipotensión ortostática y sialorrea. En los pacientes con demencia puede causar confusión temporal hasta por una semana. La clozapina se asocia con un riesgo de uno porciento de agranulocitosis y requiere de vigilancia semanal de la cuenta de leucocitos. La dosis debe ajustarse con cuidado entre 12.5 y 100 mg/día. Las dosis mayores de 150 mg/día pueden agravar los síntomas motores del Parkinson. Otros antipsicóticos atípicos que no se asocian con agranulocitosis incluyen el risperidone (0.5 a 2 mg/día) y el olanzapine (5 a 15 mg/día). Según la experiencia de los autores, el risperidone tiende a agravar los síntomas parkinsónicos con el tiempo. Por el contrario, un estudio abierto de olanzapina (5 a 15 mg/día) sugiere que este medicamento puede aliviar la psicosis inducida por medicamentos sin agravar los síntomas del Parkinson. Sin embargo, experiencias no publicadas de la institución de los autores que incluyeron a 20 pacientes sugieren que este medicamento puede no ser tan bien tolerado como la clozapina. Otros antipsicóticos alternativos que son tolerados ocasionalmente por pacientes individuales incluyen los neurolépticos de baja potencia, como la mesoridacina (10 mg/día) y el molindone (5 a 15 mg/día).
Depresión Los pacientes con depresión típicamente responden a los antidepresivos (v.gr., tricíclicos e inhibidores selectivos de la recaptura de serotonina [ISRS]). Los autores prefieren prescribir ISRS de acción corta más que de acción prolongada, ya que existen reportes de que la fluoxetina, un ISRS de acción prolongada, puede agravar la EP. Además, la combinación de fluoxetina y selegilina conlleva el riesgo teórico de un síndrome hiperserotoninérgico (delirio con mioclonía e hiperpirexia). Sin embargo, en la experiencia de los autores, no han observado ni una sola reacción hiperserotoninérgica en más de 30 pacientes tratados con esta combinación. Por último, se recomienda el tratamiento electroconvulsivo (TEC) para los pacientes con EP que sufren de depresión refractaria o que no toleran los antidepresivos orales. Existen varios reportes que indican que el TEC puede ser benéfico también para los síntomas parkinsónicos motores.
Tratamientos neuroquirúrgicos Existe nuevamente interés en el tratamiento quirúrgico del parkinsonismo intratable. En la actualidad se prefiere la palidotomía, una operación realizada por vez primera en los años 50 y después abandonada en favor de la talamotomía. Estudios clínicos recientes sobre la palidotomía han demostrado un beneficio claro en las características cardinales de la EP (acinesia/bradicinesia, temblor y rigidez), así como reducción marcada en las discinesias inducidas por medicamentos y las fluctuaciones motoras.5,50-52 Es importante no retrasar la cirugía demasiado porque el costo, en términos de discapacidad social, pérdida de independencia y autoconfianza, pérdida del empleo y deterioro en la calidad de vida, es grande y no siempre reversible. Los pacientes más jóvenes con discinesia inducida por medicamentos e intratable son los candidatos ideales. Otros tipos de tratamiento quirúrgico están actualmente en estudio (v.gr., estimulación cerebral profunda y trasplante mesencefálico fetal y de líneas celulares tratadas por ingeniería genética), y quizá demuestren ser seguros y eficaces. Los pacientes deben ser referidos a un especialista en trastornos del movimiento para ser evaluados antes de la cirugía. La presencia de demencia o atrofia cerebral importantes constituyen contraindicaciones. Además, los pacientes con formas de parkinsonismo diferentes a la EP no responden en forma favorable a la palidotomía.
OTRAS FORMAS DE PARKINSONISMO
Debido a que la tasa de errores diagnósticos en los casos de EP confirmados por autopsia es hasta del 25 porciento, es claro que otras formas de parkinsonismo pueden parecerse mucho a la EP.18 Además de satisfacer los criterios para diagnosticar la EP, es importante poner en duda el diagnóstico en las siguientes circunstancias: cuando existe historia de exposición reciente a medicamentos o toxinas que puedan causar parkinsonismo, cuando existe historia de traumatismo cerebral repetido, si los síntomas tienen inicio bilateral, si existe demencia temprana, cuando hay signos de inestabilidad postural prominente, disfunción autonómica o signos cerebelosos o piramidales. Los trastornos que se analizan a continuación son los que más se confunden con EP.
Parkinsonismo inducido por medicamentos
El parkinsonismo inducido por medicamentos es muy semejante a la EP excepto por el temblor, que suele ser (aunque no siempre) menos importante. Con frecuencia es causado por el uso subagudo o crónico de neurolépticos, carbonato de litio, antieméticos o bloqueadores de los canales del calcio.53 En una publicación europea reciente, alrededor del 30 porciento de los pacientes parkinsónicos en una clínica de trastornos del movimiento tuvieron parkinsonismo inducido por medicamentos.54 Los síntomas parkinsónicos casi siempre desaparecen días o semanas después de suspender el agente agresor. Las mujeres ancianas son especialmente propensas a sufrir este síndrome, y hasta el 40 porciento permanece afectada después de suspender el medicamento, lo que sugiere que el medicamento desenmascaró en forma prematura un fenotipo parkinsónico. Los agentes relacionados con más frecuencia han sido los bloqueadores de los canales del calcio (diltiacem y verapamil) y los antagonistas de dopamina (metoclopramida y neurolépticos).55 Sin embargo, en la experiencia de los autores, los bloqueadores de los canales del calcio causan el síndrome con mucho menos frecuencia que los antagonistas de la dopamina. Son causas menos comunes de parkinsonismo inducido por fármacos el ácido valproico y, recientemente, la fluoxetina. El parkinsonismo puede ser inducido también por la administración prolongada de agentes antihipertensivos como la reserpina, que depleta las reservas presinápticas de dopamina, y la metildopa, que bloquea la producción de dopamina. El parkinsonismo leve inducido por medicamentos puede ser aliviado por agentes anticolinérgicos, amantadina y levodopa. Los adultos jóvenes que presentan un síndrome parkinsónico deben ser interrogados respecto al uso de drogas ilegales, en espcial narcóticos sin marca que pueden ser MPTP. La exposición a sustancias tóxicas también puede causar un estado parkinsónico [ver tabla 1].
Parálisis supranuclear progresiva
La parálisis supranuclear progresiva (PSP) es uno de los síndromes parkinsónicos más comunes. Es un padecimiento diferente de etiología desconocida y que comienza típicamente en la sexta o la séptima décadas de la vida y progresa a la muerte en cinco a 10 años.55
Características clínicas Al inicio de la enfermedad son comunes las molestias sobre deterioro visual, mareo, inestabilidad, respuestas lentas, caídas y deterioro del lenguaje. El temblor es muy poco común. La expresión facial, con los ojos muy abiertos, el ceño fruncido y la cabeza hiperextendida proporciona una imagen inolvidable que contrasta con la fascies de máscara y la postura en flexión de la EP [ver figura 4]. La parálisis supranuclear característica que afecta los movimientos de los ojos hacia abajo suele ser el primer signo, seguido de deterioro de los movimientos oculares horizontales. Sin embargo, en algunos casos las características parkinsónicas son prominentes y no existen los datos oculomotores característicos, lo que puede causar un diagnóstico erróneo. El resto del examen suele revelar rigidez axial prominente (en especial de la nuca), distonía proximal y signos de motoneurona superior y cerebelosos. Un número significativo de personas con PSP puede desarrollar demencia. Se presenta degeneración diseminada y de grado variable en el tallo cerebral, los ganglios basales y el cerebelo. Aunque puede ocurrir cierta respuesta con los medicamentos antiparkinsónicos, en especial en las fases tempranas, el tratamiento no es muy eficaz.
Atrofia sistémica múltiple
Las atrofias sistémicas múltiples (ASM) son un grupo clínicamente diverso de trastornos neurodegenerativos caracterizados por diversos grados de parkinsonismo, disfunción cerebelosa y falla autonómica [ver figura 5a y b]. La edad promedio de inicio de la enfermedad son 50 años y la supervivencia promedio es de seis años.
Características clínicas El inicio es muy variable y
puede comenzar con signos de parkinsonismo, disfunción cerebelosa o
disfunción autonómica. Ocurre parkinsonismo en alrededor del 90
porciento de los casos en algún momento durante la enfermedad, y falla
autonómica en casi el 80 porciento de los casos. Los signos de falla
autonómica incluyen hipotensión ortostática,
síntomas urinarios, constipación, impotencia y pérdida de
la sudoración. Un alto porcentaje de los pacientes con ASM tiene signos
de motoneurona superior (v.gr., espasticidad y signo de
Babinsky).56
Dependiendo de las características clínicas que predominen, estos padecimientos se han agrupado en tres categorías amplias: degeneración nigroestriada, atrofia olivopontocerebelosa (AOPC) y síndrome de Shy-Drager. Los pacientes con parkinsonismo relativamente puro se clasifican como con degeneración nigroestriada. Los que tienen una combinación de parkinsonismo y otros datos, como ataxia, afección de neurona motora superior y corticobulbar, mioclonía, alteraciones oculomotoras, neuropatía periférica y sordera, se agrupan en la AOPC. Las AOPC son un grupo muy heterogéneo que incluye formas de la enfermedad tanto hereditarias como esporádicas. Solo la forma esporádica de la AOPC es parte del espectro de ASM. Si las características de parkinsonismo se asocian con signos prominentes de falla autonómica, se justifica el diagnóstico de síndrome de Shy-Drager.
Típicamente se encuentra degeneración diseminada en los ganglios basales, la médula espinal, el tallo cerebral y el cerebelo. Una característica patológica única y distintiva de la ASM es la presencia de inclusiones citoplásmicas gliales. Aún no es claro el papel de éstas en el proceso patológico.
Tratamiento En general, las características parkinsónicas de la ASM tienen una respuesta solo modesta a los medicamentos antiparkinsónicos, aunque la respuesta suele ser mejor que en la parálisis supranuclear progresiva. El tratamiento dopaminomimético en estos padecimientos suele estar limitado por la hipotensión ortostática. Ocurren discinesias inducidas por medicamentos en la ASM pero, a diferencia de la EP, suelen afectar más la cara y el cuello.
El tratamiento de la hipotensión ortostática en los pacientes con ASM (así como EP y otros trastornos asociados con hipotensión ortostática sintomática) incluye medidas simples como cambios de posición lentos, aumentar la ingesta de sal y líquidos y evitar las habitaciones demasiado calientes, las comidas abundantes y el alcohol. Otras medidas incluyen el empleo de medias elásticas, dormir en posición de Trendelenburg y ajustar otros tratamientos farmacológicos que puedan afectar la presión arterial. Debido a que la presión arterial tiende a ser mayor en la tarde, los pacientes pueden aprender a realizar la mayor parte de sus actividades cotidianas a esa hora. Si estas medidas fallan, se han propuesto diversas medida farmacológicas para tratar la insuficiencia autonómica.
Otros síntomas, como la impotencia, la urgencia urinaria y la incontinencia, así como las tormentas autonómicas con diaforesis profunsa, son más difíciles y en ocasiones imposibles de tratar por medio de fármacos. En ocasiones la impotencia mejora con la yohimbina. Los implantes de pene pueden ayudar, pero no deben realizarse sino hasta que los cambios ortostáticos estén bien controlados. La constipación puede manejarse con fibra en dosis altas, hidratación, bisacodil, concentrado de sena o lactulosa. La urgencia urinaria y polaquiuria pueden responder a la oxibutinina y al sulfato de hiosciamina; la incontinencia requiere de sondeo intermitente, uso de pañales o ambos. La gastroparesia puede mejorar con el uso de 5 a 10 mg de cisaprima antes de los alimentos; las dosis mayores pueden agravar el temblor parkinsónico.
Degeneración cortical de los ganglios basales
La degeneración cortical de los ganglios basales (DCGB) ocurre casi con tanta frecuencia como la parálisis supranuclear progresiva. El síndrome comienza típicamente en la sexta década de la vida y se manifiesta como un síndrome variado pero característicamente unilateral con rigidez, distonía, movimientos lentos y apraxia, con o sin pérdida sensorial cortical.57 Los síntomas empeoran hasta que el paciente se vuelve totalmente incapacitado. Las imágenes de resonancia magnética y patología macroscópica revelan pérdida cortical focal prominente en las áreas parietal y frontal contralateral. El examen histológico revela gliosis y neuronas edematizadas (como globos) acrómáticas con pérdida neuronal. Existen también alteraciones en la sustancia blanca que consisten en edema y desmielinización axonal, y el neuropilo tiene una apariencia espongiforme. Además de afectar la corteza, estos cambios alteran el núcleo subtalámico, el globo pálido, el nucleo estriado, el tálamo y varios nucleos cerebelosos. Los tratamientos suelen ser ineficaces.
Parkinsonismo vascular
El concepto de parkinsonismo vascular o ateroesclerótico ha sido tema de incertidumbre y controversia durante décadas. Aunque existe un acuerdo general de que esta entidad existe, su naturaleza precisa no es clara. Por lo general, los pacientes con parkinsonismo vascular muestran un síndrome aquinético-rígido sin temblor. Muchos tienen marcha de pequeños pasos.58 Además, la mayoría tiene historia de insuficiencia cerebrovascular y datos neurológicos asociados, como signos de neurona motora superior, parálisis seudobulbar y demencia, que los distingue de los pacientes con EP. Los estudios de imagen son heterogéneos y pueden revlear lagunas en los ganglios basales o infartos múltiples [ver figura 5c]. La microangiopatía hipertensiva o diabética y la enfermedad de Binswanger típicamente afecta la sustancia blanca en forma difusa o en parches confluentes en el centro semioval.58 Otras enfermedades que presentan un cuadro semejante en forma ocasional son las vasculitis inflamatorias, el síndrome antifosfolípico y, en raros casos, leucoencefalopatías como la leucodistrofia hereditaria de inicio en el adulto. Se requiere un examen de necropsia para realizar el diagnóstico definitivo debido a la ausencia de marcadores claros de la enfermedad. Desde el punto de vista terapéutico, la respuesta a los agentes dopaminérgicos es limitada, y los anticolinérgicos y la amantadina con frecuencia causan confusión y delirio.
Parkinsonismo y demencia
Existen diversas enfermedades (v.gr., enfermedad de Alzheimer [EA], de Creuzfeld-Jakob y de Pick) que tienen como datos principales tanto demencia como signos de afección de los ganglios basales. Por ejemplo, en el caso de la EA, el 25 porciento de los pacientes tienen rigidez leve y bradicinesia. Hasta el 40 porciento de los casos de EP tienen cierta patología de EA.59 Estos pacientes típicamente presentan una forma rígida aquinética de parkinsonismo, sin temblor. El curso de la enfermedad es más rápido y el tratamiento es más difícil por la alta incidencia de efectos adversos cognitivos con el tratamiento antiparkinsónico, en especial anticolinérgicos y amantadina. La toxicidad dopaminomimética central pueden presentarse de diversas formas, incluyendo trastornos del sueño (con somnolencia diurna), cambios de personalidad, depresión y torpeza mental, confusión episódica, alucinaciones y trastornos del comportamiento.
La enfermedad difusa por cuerpos de Lewy es una forma de demencia con características parkinsónicas prominentes que cada vez se detecta con mayor frecuencia.60 En algunas familias con esta enfermedad se encuentran pacientes con EP pura por enfermedad difusa por cuerpos de Lewy. Desde el punto de vista patológico, se encuentran cuerpos de Lewy en la corteza y el tallo cerebral. Algunos pacientes muestran también patología de EA, lo que dificulta la distinción de los pacientes que manifiestan un síndrome de sobreposición EA-EP. Desde el punto de vista clínico, el temblor de intención puede preceder otras características parkinsónicas, y la demencia suele ser precedida por sedación inducida por la levodopa, mioclonías y alucinaciones. Al inicio la respuesta a la levodopa puede ser importante, lo que dificulta la distinción entre enfermedad difusa por cuerpos de Lewy y EP. La progresión de los síntomas en el primer caso parece ser intermedia cuando se compaa con la progresión en la EP y los subgrupos EP-EA.
Trastornos hiperquinéticos
Los trastornos de movimiento hiperquinético incluyen diversos movimientos involuntarios (discinesias) que pueden ocurrir aislados o en combinación, e incluso en el caso de parkinsonismo. Las hiperquinesias tienen un amplio espectro de gravedad, variando de apenas detectables (v.gr., inquietud o movimiento continuo de los dedos) a violentas (v.gr., hemibalismo), e incluso a complejas y asociadas con carga emocional (v.gr., tics focales y coprolalia en el síndrome de Tourette). Cuando las discinesias ocurren en combinación, es importante determinar cuál predomina porque esto ayudará a guiar el enfoque diagnóstico y sugerir posibles tratamientos.
La corea es quizá la forma más común de discinesia.61 El término corea, que significa danza, se refiere a movimientos arrítmicos e involuntarios, que típicamente son súbitos y breves y parecen fluir de una parte del cuerpo a otra. Cuando se combinan con movimientos más lentos, como de escritura o posiciones distónicas, la discinesia se denomina coreatetosis. El hemibalismo es un tipo de corea unilateral, de gran amplitud y violento que afecta las extremidades proximales más que las distales. La causa más común de hemibalismo es una lesión vascular del núcleo subtalámico.
Son ejemplos de otros movimientos rápidos, involuntarios que pueden confundirse con corea las mioclonías, los tics y los temblores distónicos. Las mioclonías son movimientos rápidos sin la actividad rítmica que se observa en la corea. Aunque los pacientes con mioclonías con frecuencia pierden el control motor y tiran objetos, esto rara vez sucede en los pacientes con corea o tics. A diferencia de la corea y las mioclonías, los tics motores pueden suprimirse.
Los movimientos distónicos tienden a ser más lentos y las contracciones más sostenidas (segundos o más) en comparación con la corea y los tics. Al principio la distonía puede existir solo con movimientos voluntarios (distonía de acción), pero después se presenta en forma espontánea. Las contracciones dinámicas con frecuencia causan alteraciones posturales como las de la tortícolis espasmódica. Al principio los movimientos distónicos pueden responder a trucos sensoriales, como levantar un poco la barbilla para evitar que se mueva el cuello. El temblor asociado con distonía tiende a tener una calidad brusca e irregular diferente a la calidad oscilatoria de la mayoría de los otros temblores. La corea, las mioclonías, los temblores y las contracciones distónicas son percibidas por los pacientes como involuntarias. Por el contrario, los tics con frecuencia son respuestas voluntarias a neesidades subyacentes.
COREA
Etiología
La corea se ha descrito en por lo menos 150 enfermedades somáticas como una reacción a numerosos medicamentos y toxinas.61 Desde el punto de vista médico, las causas más importantes de corea son respuestas secundarias a infecciones o procesos inmunológicos, vasculares, metabólicos, endócrinos o inducidos por fármacos [ver tablas 2 y 3]. Las enfermedades sistémicas que causan corea pueden asociarse con trastornos inflamatorios, como lupus eritematoso generalizado y poliarteritis nodosa, o con infecciones o procesos parainfecciosos como corea estreptocócica, difteria, tosferina y, más recientemente, toxoplasmosis y síndrome de inmunodeficiencia adquirida (SIDA). Los trastornos cerebrovasculares que causan corea se asocian con signos neurológicos asimétricos y un curso escalonado que es compatible con un estado lacunar.
Las entidades pediátricas más importantes asociadas con corea son la corea de Sydenham, la enfermedad de Wilson, la distonía por torsión, el síndrome de Tourette y, en casos raros, la enfermedad de Huntington. Los raros casos de alteración en el metabolismo intermedio pueden causar corea, pero sus datos asociados, como encefalopatía, retraso mental y mioclonía, tienden a ser más marcados que la corea [ver tabla 4]. Los trastornos asociados con corea que pueden presentarse también en la edad adulta son la enfermedad de Wilson, la enfermedad de Huntington y la calcificación familiar de los ganglios basales (enfermedad de Fahr).
Tratamiento
Independientemente de su etiología, la corea puede tratarse con depletores de dopamina (reserpina) y bloqueadores de la misma. Se prefieren los depletores de dopamina porque tienden a producir menos efectos adversos extrapiramidales que los bloqueadores y no se asocian con el riesgo de discinesia tardía. Los efectos adversos potenciales de la reserpina incluyen parkinsonismo, distonía aguda e hipotensión postural. La reserpina inicia típicamente en dosis bajas (0.10 a 0.25 mg/día) y se aumenta según se tolere (por lo general por semana) hasta que se controla la corea o aparecen efectos adversos.
COREA DE SYDENHAM
Etiología
Conocida originalmente como mal de San Vito o corea reumática, este trastorno de la infancia y rara vez de la adolescencia, se caracteriza por corea suave, fluída y sutil asociada con hipotonía, debilidad muscular y labilidad emocional. Constituye uno de los criterios mayores de Jones para fiebre reumática y afecta a las niñas con el doble de frecuencia que a los niños, pudiendo presentarse hasta seis meses después de la infección desencadenante, con duración de entre tres a seis semanas. Es una de las secuelas posinfecciosas del estreptococo §-hemolítico. A diferencia de la corea de Sydenham, otros procesos posinfecciosos asociados con corea [ver tabla 2] típicamente se presentan como encefalopatía después de los movimientos anormales.
Manifestaciones clínicas
La corea de Sydenham típicamente no se asocia con otros datos del síndrome posestreptocócico, como carditis, poliartritis, eritema marginado y nódulos subcutáneos. La corea es antecedida por fatiga, irritabilidad, inquietud, torpeza y debilidad. Son comunes las alteraciones del comportamiento y algunos pacientes desarrollan flojera mental, depresión y ansiedad. La corea recurre varias veces durante el curso de meses en el 35 porciento de los casos. En pocas ocasiones los síntomas pueden durar varios años o incluso toda la vida. En forma periódica la corea puede reaparecer, por ejemplo durante el embarazo o después del inicio de tratamiento estrogénico.
Datos de laboratorio
Debe pensarse en la posibilidad de corea de Sydenham en cualquier niños con corea, exista o no historia de faringitis previas. El diagnóstico es apoyado por pruebas de laboratorio que indican elevación en los niveles plasmáticos de antiestreptolisina O. También pueden aumentar los niveles plasmáticos de anticuerpos menos específicos, incluyendo antiestreptocinasa, antihialuronidasa, antiestreptozima y anti-ADNasa B. Los títulos de anticuerpos antineuronales pueden estar elevados en el líquido cefalorraquídeo. En algunos casos se ha asociado la presencia de anticuerpos antineuronales circulantes con sintomatología obsesivo-compulsiva. A diferencia de los pacientes con corea vascular inflamatoria secundaria a lupus eritematoso generalizado o síndrome antifosfolípido [ver tabla 2], los pacientes con corea de Sydenham no desarrollan anticuerpos anticardiolipina.
Complicaciones
Ocurren complicaciones cardiacas agudas en alrededor del 20 porciento de los pacientes, pero pueden no ser evidentes durante la evaluación inicial. La más seria de éstas es la endocarditis, la miocarditis y la pericarditis son menos comunes. La administración profiláctica de penicilina es necesaria para prevenir las manifestaciones de fiebre reumática. Después de la corea aguda, hasta la tercera parte de los pacientes persisten con riesgo de desarrollar valvulopatía cardiaca, principalmente estenosis mitral. Para detectar esta complicación potencial en forma temprana se recomiendan los ecocardiogramas seriados.
Tratamiento
El tratamiento es estrictamente sintomático, excepto por la profilaxis con penicilina de por vida. Al inicio se recomienda reposo en cama y se prescriben benzodiacepinas o antidepresivos para controlar la ansiedad, labilidad emocional y corea. Recientemente se han usado con éxito anticonvulsivantes GABAmiméticos (GABA significa -acido gama aminobutírico), como el valproato y el gabapentin, para tratar la corea.
ENFERMEDAD DE HUNTINGTON
Etiología
La enfermedad de Huntington es un padecimiento autosómico dominante uniformemente fatal que se caracteriza por disfunción motora, emocional y cognitiva progresivas, con edad de inicio entre los 35 y los 45 años (rango de tres a 70 años).62 Es causada por el gen huntingtin, una repetición poliglutamina inestable y que se expande (CAGn) en el brazo corto del cromosoma 4.63,64 La enfermedad de Huntington existe en todo el mundo y tiene una prevalencia de 10 casos por 100,000 habitantes.
Patogenia
La neuropatología de la EH consiste en atrofia cerebral diseminada, con afección más importante en el cuerpo estriado y la corteza cerebral. Al inicio la pérdida neuronal y la gliosis son máximas en el núcleo caudado y la corteza y otras estructuras subcorticales están menos afectadas.65-67 El mecanismo de muerte celular en la EH no es claro.68 Evidencias recientes indican que la producción excesiva de poliglutamina, dirigida por el gen huntingtin anormal, es un aspecto crucial en la patogenia de la enfermedad.68-70 Desde el punto de vista experimental, ratones transgénicos que portan la expansión CAGn forman masas proteináceas, intranucleares e insolubles dentro de las neuronas de los ganglios basales y la corteza.69,70 Un fenómeno similar puede explicar las ataxias espinocerebelosas,70 un grupo de padecimientos con CAGn en otros genes.
Manifestaciones clínicas
El diagnóstico de EH puede hacerse solo por los datos clínicos en pacientes que presentan corea y tienen una historia familiar positiva. Los síntomas de demencia y emocionales (v.gr., depresión, irritabilidad) pueden precedir y típicamente ocultar los síntomas motores porque son más incapacitantes. La evolución clínica puede ser hasta de 20 años.
En las fases tempranas de la enfermedad el examen físico suele revelar movimientos coreicos (i.e., aumento en el parpadeo o gesticulación) que progresan y llegan a afectar a múltiples partes del cuerpo. La corea típicamente alcanza su máximo en 10 años y es sustituida en forma gradual por inquietud muscular, hipertonía, distonía y temblor o mioclonías.71,72 En el seis a 10 porciento de los casos la EH puede manifestar características parkinsónicas más que corea (variante de Westphal). Esta variante suele presentarse a edad más temprana (i.e., inicia en personas de menos de 20 años de edad) y muestra con mayor frecuencia un patrón de herencia paterna.73
Otras alteraciones de movimiento en la EH incluyen bradicinesia y sacadas oculares lentas. Se altera la coordinación motora gruesa y la marcha, aumenta la torpeza y los pacientes se mueven con una marcha irregular, lenta y de base ancha. La coordinación motora fina e incluso el habla se alteran en forma severa. Los trastornos en la deglución pueden ocasionar neumonía por aspiración. Los pacientes progresan hasta un estado aquinético y prácticamente mudo.
Pruebas de laboratorio
El diagnóstico de EH puede confirmarse en la actualidad con pruebas genéticas.74 No es necesario realizar estas pruebas en las personas en las que existe confirmación genética o patológica de la enfermedad en otros familiares. Las medidas diagnósticas auxiliares incluyen IRM para determinar si existe atrofia selectiva del núcleo caudado, un dato típico que puede ser difícil de detectar.
Tratamiento
El tratamiento debe incluir un equipo multidisciplinario que pueda proporcionar apoyo social, médico, neuropsiquiátrico y genético a los pacientes y familiares durante el curso de la enfermedad. Se recomienda una revisión muy completa sobre los aspectos relacionados con consejo genético y pruebas presintomáticas.74 El tratamiento sintomático puede aliviar la corea, aunque este aspecto puede no ser el más incapacitante de la enfermedad. Los bloqueadores de la dopamina tienen eficacia moderada y pueden agravar las alteraciones de movimiento no coreicas de la EH, como la bradicinesia y la distonía. Los agentes antipsicóticos atípicos como la clozapina, el risperidone y la olanzapina son mejor tolerados y pueden también ayudar a controlar la corea. Las indicaciones para tratar la corea incluyen su interferencia con las actividades de la vida diaria y sociales. El tratamiento de la depresión es semejante al de la EP.
DISTONIA
La distonía es un término empleado para referirse tanto a un síntoma como a diversos trastornos hiperquinéticos. La distonía es uno de los trastornos de movimiento más comunes y también uno de ls diagnosticados y tratados en forma equívoca con más frecuencia. La prevalencia mundial es incierta porque los casos no se notifican, pero quizá exceda 300 millones, semejante a la de la esclerosis múltiple.75
Etiología
Las distonías pueden clasificarse de acuerdo con la edad de inicio, la región afectada del organismo y la etiología. Usando un esquema etiológico semejante al usado para la EP, las distonías pueden dividirse en síndromes primarios, secundarios, con distonía asociada, y padecimientos heredodegenerativos.
Las distonías primarias incluyen síndromes en los que la distonía es la manifestación clínica principal de la enfermedad. Aunque no se ha demostrado desde el punto de vista patológico, la afección parece relacionarse con los ganglios basales. La distonía primaria es uno de los pocos trastornos de movimiento con origen en los ganglios basales que no se asocian con trastornos cognitivos o psiquiátricos primarios. Sin embargo, esto no minimiza el impacto psicológico y social de la distonía, que puede ser muy importante por la verg[Yuml]enza, ansiedad y depresión asociadas.
El principal trastorno dentro de las distonías primarias es la distonía tipo 1, un padecimiento autosómico dominante hereditario que afecta principalmente a las familias de judíos Ashkenazi (hasta en el 90 porciento de los casos). La distonía tipo 1 puede ser generalizada o focal.75 La enfermedad tiene una penetrancia de alrededor del 30 porciento y es causada por un gen localizado en el cromosoma 9q34.
La distonía que responde a dopamina (DRD) se hereda también en forma autosómico dominante, con más frecuencia en mujeres. Se caracteriza por múltiples mutaciones en el gen para la GTP ciclohidrolasa I (GTP-CH), la enzima que limita la velocidad en la síntesis del cofactor de tirosina hidroxilasa tetrahidrobiopterina.76 La tirosina hidroxilasa es la enzima que limita la velocidad de síntesis de la dopamina. La DRD típicamente inicia en la niñez, comenzando en las piernas y diseminándose a los brazos. Son comunes las fluctuaciones diurnas. Algunos pacientes inician en épocas más tardías con síntomas parkinsónicos en lugar de distonía. Las características de la DRD son una respuesta excelente a dosis bajas de levodopa y un curso de la enfermedad no progresivo. La DRD puede ser confundida con parálisis cerebral atetoide, paraplegia espástica o parkinsonismo. Aunque rara, la DRD y sus otras formas fenotípicas responden tan bien a levodopa que se justifica una prueba terapéutica en todos los casos probables. La DRD parece ser un trastorno bioquímico puro sin cambios degenerativos en el cerebro.
Las distonías secundarias, que son causadas principalmente por medicamentos o factores ambientales, incluyen la distonía inducida por levodopa, la distonía aguda y tardía asociada con bloqueadores del receptor de dopamina, la distonía asociada con parálisis cerebral (PC atetoide), el traumatismo cerebral, la hipoxia cerebral y la lesión neurológica periférica. La distonía puede asociarse también con algunos estados infecciosos y posinfecciosos y con exposición tóxica a manganeso, cianuro y ácido 3-nitropropiónico.
Los síndromas con distonía asociada incluyen algunos trastornos metabólicos de aminoácidos, lípidos y mitocondriales encefalopáticos (v.gr., encefalomielopatía necrosante subaguda [enfermedad de Leigh]).
Los padecimientos heredodegenerativos con manifestaciones distónicas secundarias se presentan típicamente con características parkinsónicas predominantes. Estos incluyen enfermedad de Wilson y de Huntington, degeneración de los ganglios basales corticales, parálisis supranuclear progresiva, la forma Lubag de distonía-parkinsonismo (distonía tipo 3) y la EP.
Manifestaciones clínicas
Los datos distónicos incluyen contracciones musculares involuntarias que pueden ocurrir durante movimientos voluntarios (distonía de acción) o en reposo, y que causan la torsión de alguna parte del cuerpo y posturas anormales. La distonía se acompaña en ocasiones de un temblor tosco e irregular (temblor distónico) o incluso de un temblor postural típico. La contracción de los músculos antagonistas es una característica fundamental de la distonía, que la distingue de la corea, los tics y otras discinesias. La distonía se asocia también con el llamado flujo exagerado, la diseminación anomal de la activación a músculos diferentes de los requeridos para el movimiento que se intenta.
Las formas más comunes de distonía son las distonías focales, que pueden afectar (1) los párpados (blefaroespasmo), causando que se cierren en forma involuntaria, (2) el cuello y los hombros (distonía cervical), causando que el cuello se tuerza a un lado (tortícolis), hacia adelante (anterocolis) o hacia atrás (retrocolis), (3) la porción inferior de la cara y la mandíbula (distonía oromandibular), y (4) la laringe (distonía espasmódica), que ocasiona una voz tirante y de calidad irregular como resultado del cierre involuntario de las cuerdas vocales. La distonía focal puede afectar las manos y antebrazos durante el desempeño de actividades específicas, como escribir (calambre del escritor), teclear o tocar un instrumento musical (calambre del músico), o durante practicamente cualquier otra actividad que incluya acciones repetitivas de las manos (distonía ocupacional). Las distonías focales suelen confundirse con problemas psiquiátricos u ortopedicos. Comparando con las generalizadas, que comienzan en la infancia, las distonías focales tienden a presentarse en etapas más avanzadas de la vida. En los niños el primer signo de distonía generalizada se presenta como una distonía activa del pie. Después la distonía ocurre en reposo y eventualmente causa alteraciones posturales al diseminarse en forma ipsilateral y después a la extremidad contralateral.
Tratamiento
El tratamiento de la distonía es estrictamente sintomático y consiste en manejo médico y quirúrgico. Los detalles del mismo van más allá de los objetivos de este capítulo. En general, para tratar la distonía generalizada se usan dosis altas de anticolinérgicos, en ocasiones en combinación con neurolépticos o depletores de dopamina. Recientemente se ha empleado con éxito la cirugía estereotáctica (palidotomía y talamotomía) para tratar casos medicamente refractarios. Las distonías focales suelen responder poco a los medicamentos, pero mucho a las inyecciones de toxina botulínica en el grupo muscular afectado. La toxina botulínica actúa bloqueando la liberación de acetilcolina en la unión neuromuscular, causando debilidad dependiente de la dosis, lo que afecta la contracción espasmódica sin producir parálisis. Se requieren inyecciones repetidas cada dos a cinco meses.
ENFERMEDAD DE WILSON
Etiología
La enfermedad de Wilson (EW), o degeneración hepatolenticular, es un padecimiento autosómico recesivo del metabolismo del cobre con una prevalencia de uno por 30,000 habitantes.77 Aunque relativamente rara, es clínicamente importante porque es tratable y simula otros trastornos del movimiento. El gen de la enfermedad de Wilson, localizado en el cromosoma 13, codifica una proteína de 7.5 kb que se expresa en gran cantidad en el hígado y el cerebro.78 La proteína pertenece a una clase de ATPasas transportadoras de cobre dependientes de ATP. Múltiples mutaciones del gen de la EW dan origen al mismo fenotipo. La mayoría de las víctimas son heterocigotos compuestos que tienen diferentes mutaciones en cada alelo. Esto complica el diagnóstico molecular de la EW y limita el escrutinio a los familiares de pacientes conocidos.79,80
Patogenia
La enfermedad de Wilson es un trastorno de la ATPasa transportadora de cobre que causa menor incorporación hepática del cobre a la ceruloplasmina, menor excreción biliar de cobre, pérdida de los sitios de almacenamiento hepático, salida de cobre hacia el plasma y depósito patológico del mismo en el cerebro (principalmente en los ganglios basales), la córnea (produciendo el anillo de Kayser-Fleisher), el riñón (causando disfunción tubular), la médula ósea (con anemia y trombocitopenia) y el sistema musculoesquelético (ocasionando artritis y osteoporosis).80 Las crisis agudas (encefalopatía aguda con crisis convulsivas, crisis hemolítica y daño tubular agudo) son causadas por salida súbita de grandes cantidades de cobre hacia el torrente sanguíneo por iatrogenia (ver adelante) o después de infartos hepáticos o hepatitis aguda.
Manifestaciones clínicas
La edad de presentación varía entre tres y 60 años, con un pico de inicio alrededor de los 16 años. En un estudio de una serie extensa, el 54 porciento de los pacientes presentó manifestaciones neurológicas, 31 porciento disfunción hepática y hasta el 33 porciento manifestaciones psiquiátricas.77 La mayoría de los pacientes con alteraciones hepáticas son mujeres entre los ocho y los 16 años de edad, los hombres presentan con más frecuencia alteraciones neurológicas. El inicio puede variar desde falla hepática aguda fulminante hasta cirrosis insidiosa y leve. La EW puede ser la causa más común de disfunción hepática crónica en niños; todos los niños con esta manifestación deben ser estudiados buscando EW. Las principales manifestaciones hematológicas de la enfermedad incluyen anemia hemolítica, trombocitopenia (50 porciento de todos los pacientes con manifestaciones hematológicas) y neutropenia (30 porciento). Otras manifestaciones sistémicas son alteraciones del páncreas exócrino, hipoparatiroidismo, poliartritis, cálculos renales y cardiomiopatía por acúmulo miocárdico de cobre.
Las manifestaciones neurológicas de la EW son muy diversas, incluyendo varios trastornos del movimiento como distonías (65 porciento de los pacientes), rigidez o parkinsonismo (52 porciento), alteraciones de la postura y la marcha (42 porciento) y temblor (32 porciento). Sin embargo, la manifestación neurológica más común es la disartria (97 porciento), en ocasiones asociada con expresiones faciales alteradas (la clásica risa sardónica). Las manifestaciones psiquiátricas pleomórficas y de comportamiento de la EW incluyen irritabilidad, menor desempeño laboral, depresión y cambios en el estado de ánimo. Los trastornos afectivos pueden simular enfermedad bipolar y ocurren también síntomas psicóticos. Los problemas cognitivos pueden iniciar con pobre desempeño escolar y culminar en demencia.
Debido a la extrema variabilidad de las manifestaciones neuropsiquiáticas en la EW, es importante pensar en este diagnóstico en prácticamente todos los trastornos del movimiento que se presentan sin una etiología demostrable, en especial en jóvenes con sintomatología psiquiátrica.
Alteraciones de laboratorio
El diagnóstico puede hacerse en alrededor del 80 porciento de los casos al demostrar reducción en la ceruloplasmina sérica (EW, 0 a 200 mg/L, normal de 200 a 400 mg/L).77,81 Los pacientes con niveles de seruloplasmina normal se consideran heterocigotos. También está disminuido el cobre sérico (unido más libre), (EW 3 a 100 µmol/l, normal 11 a 24 µmol/L). La excreción aumentada de cobre en orina de 24 horas puede ser una medición más sensible que los niveles en suero (EW 100 a 1,000 µg en 24 horas, normal ² 40 µg en 24 horas), pero con este resultado, debe distinguirse a la EW de otros trastornos que producen colestasis. En los casos en los que los datos anteriores no son concluyentes, la prueba definitiva es la demostración de mayor contenido de cobre en la biopsia hepática (EW 200 a 3,000 µg/g de tejido seco, normal ² 50 µg/g de tejido seco). Casi todos los pacientes que tienen afección neurológica tienen los datos oftalmológicos clásicos del anillo de Kayser-Fleischer, o cataratas en girasol. Estos anillos representan depósitos de cobre en la membrana basal de la córnea [ver figura 6], y su detección se realiza con lámpara de hendidura. Los estudios de neuroimagen revelan pérdida del volumen cerebral, con dilatación ventricular en más del 95 porciento de las personas con afección neurológica, así como algunas neuropsiquiátricamente intactas. La IRM revela una señal hiperintensa única en las imágenes en T2 del núcleo lenticular, así como en las imágenes del tálamo, el puente mesocerebral y la circunvolución dentada en pacientes con enfermedad más avanzada [ver figura 5d]. Las imágenes en T1 pueden revelar lesiones de la sustancia blanca.82
Tratamiento
El tratamiento debe ser cuidadoso para evitar la encefalopatía aguda o la toxicidad hepática inducida por una movilización rápida del cobre.83,84 Los autores realizan una IRM antes de iniciar el tratamiento, incluso en pacientes asintomáticos. Este consiste en disminuir el cobre de la dieta y disminuir su absorción con acetato o sulfato de zinc (150 mg/día) o sulfuro de potasio (20 mg tid). Para movilizar los depósitos de cobre en los tejidos es necesario usar quelación del mismo con penicilamina. En los pacientes que no toleran la penicilamina (1 a 3 g/día por vía oral) o en los que empeoran al iniciar el tratamiento (10 a 40 porciento), otras alternativas incluyen el uso de trientine (con o sin dosis bajas de penicilamina) o dimercaprol y cloruro de cobalto (que rara vez se usa en la actualidad). Otras estrategias para disminuir el daño a los tejidos por el depósito de cobre son el uso de depuradores de radicales libres, como el mercaptodextrán.
El tratamiento quelante requiere vigilancia estrecha para lograr una excreción inicial de cobre en orina de 24 horas de 1.2 a 2.0 g/día. El tratamiento con penicilamina es mal tolerado en hasta el 30 porciento de los pacientes por efectos adversos alérgicos idiosincráticos, incluyendo condiciones autoinmunes. Contrario a la penicilamina, el tratamiento con zinc es bien tolerado. Puede aumentar la amilasa, lipasa y fosfatasa alcalina, y existen reportes aislados de efectos inmunosupresores. Cuando existe evidencia de progresión de la enfermedad a pesar del tratamiento dietético y farmacológico óptimos o cuando ocurre falla hepatica, el trasplante autólogo de hígado es el tratamiento más definitivo.85
Los síntomas neurológicos suelen responder mal al tratamiento. Pueden administrarse anticolinérgicos para tratar la distonía y el temblor, y administrarse dopaminomiméticos para la rigidez. Los síntomas psiquiátricos (ansiedad y depresión) se tratan en la forma habitual. La mejoría en los síntomas neuropsiquiátricos y otros puede retrasarse por meses o hasta por dos años después de iniciado el tratamiento quelante.
SINDROME DE TOURETTE
Etiología
El síndrome de Tourette (ST) es un padecimiento autosómico dominante en el que no se ha identificado el gen responsable. Tiene una expresión fenotípica muy amplia, penetrancia variable y expresión de género. Los criterios diagnósticos incluyen la presencia de múltiples tics motores y vocales que duran más de un año y aparecen antes de los 21 años. Los síntomas pueden ser suficientemente severos para justificar un diagnóstico médico y la alteración puede no asociarse con otros trastornos neurológicos o inducidos por medicamentos. Es importante la historia familiar, pero puede ser enmascarada por el fenotipo, que incluye sintomatología obsesivo-compulsiva (SOC) y trastornos de atención deficiente. La relación hombre-mujer para los tics es de 4:1, y la de la SOC es de 1:4. También pueden existir trastornos del aprendizaje y varios síndromes de comportamiento mal definidos.
Patogenia
La patogenia del síndrome de Tourette se desconoce, pero la evidencia farmacológica clínica y estudios limitados de autopsia sugieren que puede ser mediada por innervación hiperdopaminérgica del cuerpo estriado. Estudios morfométricos recientes que usan IRM del cerebro y estudios metabólicos usando tomografía de emisión de positrones sugieren la presencia de alteraciones anatómicas y funcionales en los circuitos corticoestriadopalidotalámico que se proyectan a la corteza prefrontal y a la circunvolución del cíngulo. La interrupción quirúrgica de estos circuitos (v.gr., tractotomía anterior y cingulotomías) puede ser útil para tratar la SOC y, en menor grado, los tics en casos seleccionados.
Manifestaciones clínicas
Un tic es una acción breve, estereotipada, rápida, repetitiva y aparentemente sin propósito que incluye uno o varios grupos musculares. Los tics tanto motores como vocales pueden ser simples (v.gr., parpadeo frecuente, retorcerse la nariz, toser, gruñir) o complejos (automutilación y coprolalia). Los tics pueden asociarse con experiencias sensoriales focales (tics sensoriales), que pueden preceder, acompañar o seguir a los movimientos o vocalización. En general los tics son la parte menos incapacitante del ST y pueden no requerir tratamiento. Durante los años escolares los trastornos en la atención, comportamiento (impulsividad e irritabilidad) y aprendizaje tienden a ser más incapacitantes. La SOC comienza al inicio de la adolescencia y puede ser muy incapacitante. Responde a clomipramina y a inhibidores selectivos de la recaptura de serotonina con menos frecuencia que el trastorno obsesivo-compulsivo sin ST. Los trastornos del sueño son frecuentes, pero mal comprendidos.
Tratamiento
El tratamiento del ST es difícil y debe realizarse por especialistas. Cuando los síntomas son complejos (v.gr., tics, SOC y deficiencia en la atención) los pediatras y neurólogos deben actuar junto con psiquiatras que guien y traten los síntomas de comportamiento. Los tics suelen responder a bloqueadores de dopamina (v.gr., prolixina, haloperidol y pimozide). Algunos niños son especialmente intolerantes a los efectos adversos de estos medicamentos, que varían de distonía aguda e inquietud motora (acatisia) a sedación diurna y fobias escolares. Reportes recientes sugieren que el neuroléptico atípico risperidone puede ser eficaz. Otros tratamientos para los tics incluyen clonidina (0.1 a 0.4 mg/día, como una cápsula o parche transdérmico) y clonacepam (0.5 a 4.0 mg/día).
Temblor fisiológico y esencial
El término temblor se usa para describir varios movimientos no relacionados que comparten una característica en común: oscilación de una extremidad o parte del organismo. Son ejemplos el templor en reposo del Parkinson y el temblor cerebeloso.
INCREMENTO DEL TEMBLOR FISIOLOGICO
El temblor fisiológico típicamente no es aparente, excepto cuando es exagerado por estrés, ansiedad o estimulantes. El miedo y otras formas de ansiedad situacional son ejemplos bien reconocidos y tratables de temblor fisiológico exagerado. Otras causas posibles de incremento del temblor incluyen agentes adrenérgicos, como la liberación endógena de epinefrina (por feocromocitoma, miedo o hipoglucemia) y liberación exógena de epinefrina, isoproterenol, terbutalina y teofilina. Los medicamentos que pueden inducir temblor fisiológico incluyen al litio, antidepresivos tricíclicos, estimulantes, valproato, esteroides, hormonas tiroideas y cafeína. La supresión de ciertos fármacos, como el alcohol, los opiáceos y las benzodiacepinas, así como el ejercicio excesivo (creando fatiga muscular) pueden causar mayor temblor fisiológico.
Tratamiento
El tratamiento incluye la eliminación o reducción de cualquiera de los agentes desencadenantes del temblor por medio de bloqueadores beta adrenérgicos (v.gr., propranolol), controlando la ansiedad y, cuando está indicado, tratando la endocrinopatía asociada. La ansiedad situacional puede controlarse administrando propranolol (20 a 60 mg) una hora antes del evento o situación desencadenante.
TEMBLOR ESENCIAL
El temblor esencial (TE) tiene una frecuencia de 6 a 8 Hz, es de origen central y con frecuencia tiene un patrón de herencia autosómico dominante.87 A diferencia de los pacientes con EP, los enfermos con TE pueden no tener otras características parkinsónicas y presentar solo movimiento aislado de la cabeza y temblor en la voz. Este último da al lenguaje una calidad vacilante. Los síntomas del TE se exacerban por los mismos factores que aumenta el temblor fisiológico.
Tratamiento
Característicamente, el temblor esencial responde rápido aunque en forma breve al alcohol (sin embargo, al disminuir la concentración sérica de alcohol el efecto de rebote puede empeorar el temblor). También responde a bloqueadores adrenérgicos beta (v.gr., propranolol) que suelen ser eficaces a largo plazo. Otros betabloqueadores útiles incluyen metoprolol, timolol, atenolol y sotalol. La primidona puede ser eficaz en dosis de 50 a 250 mg/día, que son menores que las requeridas para el tratamiento anticonvulsivante. Otros medicamentos que pueden ser benéficos incluyen benzodiacepinas (en especial clonacepam), fenobarbital y clonidina. La neurocirugía estereotáctica (talamotomía ventrolateral o estimulación cerebral profunda) es útil para los temblores severos refractarios a medicamentos.
Mioclonías
Características clínicas
Las mioclonías son movimientos súbitos, breves, involuntarios, como descargas. Pueden considerarse fragmentos de epilepsia, y tienen características electroencefalográficas que son semejantes a las descargas epilépticas focales. Las mioclonías positivas consisten en contracción muscular activa y las negativas en un lapso súbito dentro de la contracción que es clínicamente indistinguible de la asterixis. Las pruebas electrofisiológicas especializadas pueden localizar las descargas mioclónicas en la corteza somatosensorial, el tallo cerebral (el sistema activador reticular ascendente) y, en casos raros, la médula espinal. Como las crisis convulsivas, las descargas mioclónicas representan un signo específico de irritación del SNC con un diagnóstico diferencial amplio [ver tabla 4].86
Las descargas mioclónicas pueden ser focales, segmentarias o generalizadas. Cuando son focales sugieren una patología focal, que varía de epilepsia a encefalitis focal [ver tabla 4]. Pueden ser espontáneas, sensibles a estímulos (reflejas) o relacionarse a actividades (de acción). Cuando son generalizadas pueden asociarse con epilepsia o con una encefalopatía estática y progresiva. Para disminuir las posibilidades diagnósticas durante el examen es importante buscar signos de encefalopatía, demencia, patología retininana, pérdida de la audición, miopatía, neuropatía y ataxia.
Diagnóstico diferencial
El diagnóstico diferencial de las mioclonías consiste en determinar si las contracciones son causadas por una enfermedad médica, exposición a un fármaco o toxina, o un trastorno primario del sistema nervioso central.89 Un elemento importante en el diagnóstico diferencial es si las mioclonías son epilépticas o no.90,91 La edad de inicio es muy importante porque las formas progresivas más comunes de epilepsia que son causadas por procesos degenerativos tienden a ocurrir en el grupo de edad pediátrico.
Pruebas de laboratorio
El escrutinio de laboratorio básico debe incluir una biometría hemática, química sanguínea y estudio toxicológico. Están indicados los estudios de imagen cerebral y la electroencefalografía cuando la causa de la mioclonía no es inmediatamente aparente. Cuando la clínica lo justifica deben realizarse estudios metabólicos especializados para descartar trastornos inflamatorios sistémicos, endocrinopatías, trastornos del metabolismo del cobre (v.gr., EW y enfermedad de Menke), deficiencias de enzimas seleccionadas y trastornos de almacenamiento de lisosomas [ver tabla 4 ]. Un subgrupo de estos padecimientos, como algunos de los trastornos de la fosforilación oxidativa mitocondrial (ver adelante) y algunas de las ataxias espinocerebelosas pueden diagnosticarse en la actualidad con pruebas genéticas.
Tratamiento
El tratamiento de las mioclonías es estrictamente sintomático. Los medicamentos de uso común incluyen clonacepam (2 a 12 mg/día), valproato de sodio (600 a 3,000 mg/día), primidona (500 a 1,500 mg/día) y, en Europa, piracetam (6 a 20 mg/día). Los inhibidores selectivos de la recaptura de serotonina y el 5-hidroxitriptofano (con carbidopa) pueden ayudar en algunas formas específicas de mioclonía (la mioclonía reticular refleja) pero puede agravar otras. Otra clase especial es la mioclonía del paladar, que responde a trihexifenidilo (6 mg/día). El tratamiento de los síndromes epilépticos asociados con trastornos mioclónicos no será analizado en este capítulo.
MIOCLONIAS EN LA PRACTICA MEDICA GENERAL
En la práctica médica general la forma más común de mioclonía es resultado de trastornos metabólicos, en los que las mioclonías negativas o asterixis tienden a dominar el cuadro clínico [ver tabla 4]. La etiología de las mioclonías incluye también encefalitis viral, que típicamente se presenta como mioclonías positivas y negativas. Son ejemplos las encefalitis asociadas con virus herpes simple y arbovirus, así como las asociadas con síndromes posinfecciosos (v.gr., panencefalitis esclerosante subaguda y encefalitis letárgica).
Otras formas comunes de mioclonías observadas en la práctica médica general incluyen mioclonías inducidas por medicamentos y mioclonías secundarias a suspensión de fármacos. Los medicamentos que inducen mioclonías incluyen carbonato de litio, antidepresivos tricíclicos e inhibidores selectivos de la recaptura de serotonina (ISRS). La coadministración de ISRS e inhibidores de la MAO A o MAO B pueden causar un síndrome hiperserotoninérgico que típicamente inicia con mioclonías y cambios en el estado mental. Los agentes antinfecciosos asociados en ocasiones con mioclonías incluyen a las penicilinas y las cefalosporinas [ver tabla 3]. En la anestesia, los hipnóticos no barbitúricos de acción corta (v.gr., etomidato) pueden causar también mioclonías. Los pacientes con EP que están tomando levodopa en ocasiones desarrollan mioclonías.
La sobredosis o intoxicación con anticonvulsivantes, antihistamínimos o hipnosedantes pueden causar mioclonías, con o sin crisis generalizadas. Otros medicamentos incluyen los antineoplásicos como el clorambucil, los antinflamatorios no esteroides (AINE) cuando se administran a pacientes con insuficiencia renal y el bismuto. La exposición extensa a las siguientes toxinas también se asocia con mioclonías: metilbromuro, mercurio orgánico, tetraetilo de plomo (gasolina), estricnina y DDT. La supresión de alcohol, benzodiacepinas u opiáceos causa mioclonías con frecuencia. Por último, las mioclonías pueden ser una secuela de la lesión cerebral global por traumatismos, anoxia-isquemia, golpe de calor y electrocución.92
Los trastornos demenciales asociados con mioclonías incluyen padecimientos asociados a priones, como la enfermedad de Creutzfeld-Jakob. Además, un pequeño subgrupo de pacientes con enfermedad de Alzheimer (10 a 15 porciento) y un gran número de pacientes con enfermedad difusa de cuerpos de Lewy presentan mioclonías. Estos padecimientos pueden evolucionar de mioclonías focales a formas más generalizadas.93
Algunos otros trastornos representan problemas genéticos primarios poco comunes con sintomatología compleja asociada [ver tabla 4].94,95 Además de controlar las mioclonías y crisis consulsivas, el tratamiento para estas condiciones es muy limitado.
Trastornos del movimiento inducido por medicamentos
Los trastornos del movimiento inducidos por medicamentos son un grupo importante de condiciones iatrogénicas y casi siempre reversibles que se presentan con frecuencia en la práctica clínica general. La frecuencia de los trastornos del movimiento inducidos por medicamentos dependen de la naturaleza del compuesto, la edad de la persona afectada y otros factores de riesgo (ver adelante).96
La mayoría de los trastornos del movimiento relacionados con medicamentos se asocian con agentes que afectan en forma directa o indirecta la trasmisión dopaminérgica central. Las categorías de fármacos incluyen los estimulantes del SNC, el precursor de dopamina levodopa, los agonistas dopaminérgicos de acción directa y los bloqueadores del receptor central de dopamina. Los tipos de movimientos producidos por estos compuestos pueden ser agudos, subagudos o tardíos, como en el caso de la discinesia tardía o distonía tardía. Los mecanismos de los trastornos de movimiento agudos y subagudos parecen ser extensiones idiosincráticas de la acción del compuesto. Por el contrario, el mecanismo de la discinesia tardía no se ha aclarado.
Alrededor de la tercera parte de los pacientes con discinesia tardía remiten a los tres meses de suspender el tratamiento neuroléptico. En la mayoría de los pacientes los movimientos remiten en forma gradual en cinco años. Los pacientes con riesgo de discinesia tardía permanente incluyen los ancianos, los desdentados y los que sufren disfunción orgánica cerebral. Los pacientes con enfermedades afectivas tienen más riesgo de desarrollar discinesia tardía que los esquizofrénicos.
TRASTORNOS DEL MOVIMIENTO AGUDOS INDUCIDOS POR FARMACOS
Las reacciones de esta categoría incluyen la distonía aguda en respuesta a bloqueadores centrales de dopamina. Parecen ser más frecuentes en varones jóvenes y en pacientes que reciben medicamentos como litio, bloqueadores de los canales del calcio e ISRS. La distonía aguda puede tratarse con la administración parenteral de anticolinérgicos (benzotropina o difenhidramina) o benzodiacepimas (loracepam o diacepam). Otros trastornos de movimiento agudos incluyen discinesias, comportamientos estereotipados en respuesta a neurolépticos y tics, que pueden ocurrir en respuesta a estimulantes del SNC como metilfenidato, dextroanfetamina y pemoline.
TRASTORNOS DEL MOVIMIENTO SUBAGUDOS INDUCIDOS POR FARMACOS
Es probable que la reacción más común de este tipo sea la acatisia inducida por neurolépticos.97 La acatisia es un estado de inquietud motora que consiste en una necesidad imperiosa de moverse. El movimiento tiende a aliviar los síntomas en forma temporal. La inquietud parece estar mediada por el bloqueo del receptor de dopamina mesolímbico. Desde el punto de vista sintomático, esto puede relacionarse con el síndrome de piernas inquietas. El tratamiento consiste en retirar el agente agresor. Cuando esto no es posible los síntomas pueden disminuir con benzodiacepinas, anticolinérgicos, betabloqueadores y, en algunos casos, agentes dopaminérgicos.
TRASTORNOS DE MOVIMIENTO TARDIOS
Manifestaciones clínicas
Los trastornos de movimiento tardíos son causados principalmente por exposición crónica (mayor de tres meses) a bloqueadores centrales de dopamina.98 Los movimientos son principalmente coreicos y se conocen como discinesia tardía (DT). Al inicio se afectan los músculos del cráneo (boca, labios y lengua en particular), y después el tronco y las extremidades. En los casos más avanzados una persona puede sufrir movimiento continuo de la cabeza, rotación de la pelvis y movimientos finos de los dedos de manos y pies. En raros casos la discinesia tardía puede afectar el diafragma y causar falla respiratoria. Los pacientes con distonía tardía presentan contracciones distónicas axiales principalmente, más que apendiculares. Otros síndromes tardíos incluyen la acatisia tardía, los tics tardíos e incluso temblor tardío.99
Tratamiento
El mejor tratamiento para la DT es la prevención. Se recomienda el uso juicioso de neurolépticos por el periodo más corto posible. Los neurolépticos atípicos se asocian con un riesgo significativamente menor de causar DT que los neurolpépticos convencionales, para los que el riesgo varía de 13 a 20 porciento. El menor riesgo asociado a los neurolépticos atípicos puede atribuirse a su mayor afinidad por los receptores de dopamina mesolímbicos D4 y D3, más que los receptores estriados D2. Los neurolépticos atípicos incluyen la clozapina, el resperidone y la olanzapina, recién autorizada.
Cuando no es posible suspender el agente que causa la DT, debe considerarse cambiar los neurolépticos tradicionales por atípicos.99 Si el paciente está tomando también estimulantes y anticolinérgicos, el eliminarlos puede aliviar la discinesia. La reserpina es el tratamiento de elección para la discinesia tardía coreiforme. La dosis inicial debe ser de 0.125 mg/día y puede aumentarse lentamente hasta 6 mg/día, según se requiera. Los ancianos tienen menos probabilidad de tolerar el tratamiento por hipotensión ortostática. El uso a largo plazo de reserpina se asocia también con un incremento de 15 porciento en la incidencia de depresión.
Otra estrategia de tratamiento consiste en estimular el sistema GABAérgico usando baclofen (40 a 80 mg/día), clonacepam (1 a 8 mg/día) o ácido valproico (1 a 3 g/día). Las estrategias GABAérgicas son especialmente útiles en pacientes con discinesia tardía que pueden beneficiarse del tratamiento anticolinérgico y de inyecciones de toxina botulínica en los músculos más afectados. La buspirona (40 a 60 mg/día) es un tratamiento GABAmimético que ha demostrado ser útil en algunos casos.
Reconocimientos
Este capítulo fue patrocinado en parte por una beca del Centro de Excelencia de la Emory University School of Medicine de la American Parkinson's Disease Association, de Nueva York.
Figuras 1 y 2 Seward Hung
Figura 3 Cortesía del Dr. Thomas M. Aaberg, Sr., Servicio de Oftalmología, Emory University School of Medicine.
DR. JORGE L. JUNCOS
DR. MAHLON R. DELONG
La mayoría de los trastornos de movimiento se deben a enfermedades que afectan los ganglios basales. Los trastornos hipoquinéticos se caracterizan por alteración de los movimientos voluntarios, el parkinsonismo es un ejemplo. Los trastornos hiperquinéticos se presentan como diversos síndromes clínicos, incluyendo corea, balismo, distonía, temblor, tics y mioclonías.
Cada trastorno de movimiento típicamente tiene múltiples etiologías pero fisiopatología y fenotipo compartido. En ausencia de marcadores biológicos y patrones de herencia claros, el diagnóstico etiológico de la mayoría de los trastornos de movimiento es un ejercicio clínico muy difícil. Sin embargo, los adelantos importantes en la comprensión de la organización funcional de los ganglios basales y en la fisiopatología de los trastornos del movimiento ha originado el desarrollo de nuevos enfoques farmacológicos y quirúrgicos para el tratamiento.
Los trastornos de movimiento rara vez son trastornos solo del movimiento. Las alteraciones psiquiátricas y cognitivas asociadas pueden contribuir en forma significativa e incluso dominar en el cuadro clínico. Es importante detectar la morbilidad psiquiátrica concomitante porque ésta puede contribuir en forma desproporcionada al trastorno. Por ejemplo, la aparición de demencia y depresión puede causar deterioro clínico inexplicable a pesar del tratamiento adecuado de los síntomas motores.
Fisiopatología de los trastornos del movimiento
Los trastornos de movimiento que se originan en los ganglios basales parecen depender de disfunción del circuito motor ganglios basales-tálamocortical [ver figura 1].1 Las alteraciones de las señales de salida de los ganglios basales a nivel del segmento interno del globo pálido (GPi) puede causar trastorno en los movimientos tanto voluntarios como involuntarios.23
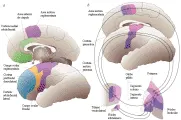
|
| Figura 1 |
| Anatomía funcional de los ganglios basales |
Se ha desarrollado un modelo fisiopatológico de parkinsonismo con base en estudios animales [ver figura 2]. La característica bioquímica de este síndrome es la deficiencia de dopamina, que promueve una actividad excesiva de la llamada vía indirecta. Esto aumenta el estímulo excitatorio sobre el GPi desde el núcleo subtalámico.2 La mayor influencia inhibitoria del GPi ocasiona mayor inhibición de las neuronas tálamocorticales, lo que provoca que algunas áreas de proyección cortical tengan menos respuesta a estímulos que en condiciones normales participan en el inicio y ejecución del movimiento. Por ejemplo, la aquinesia y la bradicinesia son causadas por inhibición excesiva de las neuronas tálamocorticales. El apoyo para este modelo deriva de estudios que demuestran que la inactivación selectiva del núcleo subtalámico o GPi reduce mucho los signos motores de la enfermedad de Parkinson (EP), incluyendo la aquinesia.4,5 Además, se ha demostrado el restablecimiento en pacientes de la activación cortical durante una tarea de movimiento por medio de mediciones en tomografía de emisión de positrones (TEP) antes y después de la palidotomía, un procedimiento que destruye quirúrgicamente el segmento motor del GPi.6
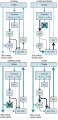
|
| Figura 2 |
| Flujo estriado y los trastornos del movimiento |
En contraste con los trastornos hipoquinéticos, los hiperquinéticos son resultado de un estímulo disminuido del GPi [ver figura 2].7 En este caso las neuronas tálamocorticales, que no tienen el impulso tónico inhibidor normal proveniente del GPi, se vuelven más sensibles a los estímulos corticales o muestran una mayor tendencia a descargar en forma espontánea, lo que ocasiona movimientos deteriorados o involuntarios. Tanto en el hemibalismo, que es causado de lesiones del núcleo subtalámico, como en la enfermedad de Huntington (EH), en la que ocurre una pérdida selectiva temprana de las neuronas estriadas de la vía indirecta, la menor salida de estímulos del núcleo subtalámico causa actividad anormalmente baja del GPi y menores niveles de inhibición talámica.8
Existen evidencias contundentes acerca de que los trastornos hipo o hiperquinéticos que clínicamente son contrastantes, son causados en forma respectiva por niveles excesivos o reducidos de estímulos del GPi del circuito motor, con inhibición o desinhibición talámica que ocasiona los síndromes respectivos. Cambios fisiopatológicos semejantes son responsables de los trastornos de los movimientos oculares que ocurren después del daño al circuito oculomotor tanto en la EP como en la EH. Es tentador especular sobre si ocurren cambios patológicos comparables en otros circuitos ganglios basales-tálamo para producir los complejos trastornos de comportamiento, cognitivos y límbicos equivalentes de los síntomas y signos motores que ocurren en la disfunción del circuito motor (v.gr., los síndromes del lóbulo frontal, el pensamiento lento [bradifrenia], la manía y la depresión, los tics y los trastornos obsesivo-compulsivos).9
Trastornos hipoquinéticos
El parkinsonismo es un síndrome clínico con múltiples etiologías que se caracteriza por grados variables de discinesia (movimiento lento), temblor, rigidez y inestabilidad postural.10 El común denominador de todas las formas de parkinsonismo es la reducción de la trasmisión dopaminérgica estriada como resultado de disrupción estructural o funcional de las vías nigroestriadas. El parkinsonismo puede dividirse en enfermedad de Parkinson idiopática, que es responsable de alrededor del 75 porciento de los casos, y formas secundarias de parkinsonismo [ver tabla 1]. Es importante distinguir la EP de otras formas de parkinsonismo porque el pronóstico y tratamiento son diferentes. El diagnóstico temprano y adecuado se vuelve incluso más importante cuando existen tratamientos que pueden retrasar o detener la progresión del proceso patológico subyacente.
|
||
|
ENFERMEDAD DE PARKINSON IDIOPATICA
Etiología
La enfermedad de Parkinson es un trastorno neurodegenerativo de etiología incierta que afecta a más de un millón de personas en los Estados Unidos, alrededor de uno porciento de los mayores de 55 años.11 Es responsable de casi tres cuartas partes de los casos de parkinsonismo. Los papeles relativos de los factores hereditario, ambiental y endógeno son inciertos. Varios estudios epidemiológicos y familiares recientes indican que la historia familiar positiva, cuando se investiga en forma adecuada y detallada, es mucho más común de lo que se pensaba antes. En parte con base en la revisión de datos obtenidos de estudios en gemelos monocigotos, en la actualidad parece ser que los pacientes con EP pueden heredar una vulnerabilidad genética para la enfermedad.12 Sin embargo, la manifestación del fenotipo depende del proceso de envejecimiento (v.gr., pérdida relacionada con la edad de la capacidad de fosforilación oxidativa) y de una serie de otros factores de desarrollo, ambientales y genéticos no identificados. Los estudios epidemiológicos han demostrado que el riesgo de EP es mayor entre quienes viven en áreas rurales, beben agua de pozo o están expuestos a pesticidas.13 El consenso actual es que es muy probable que la EP sea causada por una interacción entre factores genéticos, ambientales y estrés oxidante endógeno.
En un artículo clave, una mutación en el gen alfa-sinucleína del cromosoma 4 se asoció con una forma autosómico dominante de EP en un pedigree italiano extenso y en tres familias no relacionadas de origen griego.14 La mutación consiste en la sustitución de una base en la posición 209 (G209T). El gen codifica la proteína precursora del componente amiloide no § de las placas neuríticas de la enfermedad de Alzheimer. La proteína alfa-sinucleína se encuentra en las terminales presinápticas y su distribución en el cerebro es semejante a la de los cuerpos de Lewy (la presencia de inclusiones citoplasmáticas prominentes).14 Se ha postulado que la proteína mutada puede tener una estructura terciaria anormal, lo que la hace propensa a autoagregarse y a formar núcleos semejantes a amiloide, como los encontrados en los cuerpos de Lewy y en las placas dendríticas.14 Aunque la mayoría de los casos no son autosómico dominantes, este descubrimiento puede orillar a un nuevo conocimiento fundamental sobre la fisiopatología de la muerte de las células dopaminérgicas, que ocurre en todos los pacientes con EP.
Patogenia
Las características patológicas de la EP son la degeneración de las células dopaminérgicas de la sustancia negra compacta (SNc) y de los cuerpos de Lewy en las neuronas pigmentadas del tallo cerebral, incluyendo el locus ceruleus, el núcleo del rafé pontino el núcleo motor dorsal del nervio vago.15 Aunque la depleción celular en la SNc y otras áreas ocurre en forma gradual durante muchos años, los síntomas se desarrollan solo cuando se han perdido entre el 50 y el 80 porciento de las células y han fallado los mecanismos compensatorios.
Se ha puesto gran atención al papel del estrés oxidativo en la EP y al daño que pueden causar los radicales libres producidos por el metabolismo de la dopamina y otras sustancias.16 La disfunción mitocondrial es otro factor posible que puede contribuir a la vulnerabilidad al estrés oxidativo, y se ha reportado evidencia de disfunción mitocondrial en el músculo y las plaquetas de los pacientes con EP.17 En la actualidad, el papel de la disfunción mitocondrial en la EP es un punto de gran interés y controversia.
Características clínicas
Lo más común es que la EP se desarrolle en forma insidiosa hacia la mitad de la vida, con un pico en la edad de inicio entre los 55 y los 65 años [ver figura 3]. En los estadios iniciales de la enfermedad el diagnóstico puede ser difícil, a menos que exista el temblor en reposo característico de la enfermedad. En ausencia de temblor, el diagnóstico puede pasar desapercibido si la atención se enfoca en lo que parece debilidad, molestias musculoesqueléticas inespecíficas o problemas neuromusculares periféricos. Las características clínicas de la EP son bradicinesia, rigidez muscular y un temblor en reposo de 4 a 5 Hz. La presencia de dos de estos tres signos cardinales y una respuesta clínica clara a la levodopa, el precursor de la dopamina, son suficientes para hacer el diagnóstico de probable EP.18 El inicio unilateral de los síntomas y la presencia de temblor en reposo favorece en forma intensa el diagnóstico de EP. La fascies inexpresiva, el menor parpadeo, la postura inclinada, los movimientos lentos y la micrografia (escritura que se deteriora y disminuye de tamaño) suelen ser frecuentes en las fases tempranas de la enfermedad.

|
| Figura 3 |
| Cara de un paciente con Parkinson. |
La bradicinesia, o retraso en la ejecución de movimientos, y los movimientos escasos (acinesia) son de las características más incapacitantes de la EP porque interfieren con todos los aspectos de la vida diaria, incluyendo actividades como caminar, levantarse de una silla, voltearse en la cama o vestirse. También se afecta el control fino, que se manifiesta por menor destreza de los dedos y micrografía. La hipofonía y el lenguaje monotónico son también manifestaciones de bradicinesia.
El temblor en reposo comienza típicamente en la porción distal del brazo, en donde se presenta como un movimiento constante de «frota monedasÕ. El temblor suele progresar en dirección proximal en el brazo y después en la pierna antes de cruzar hacia el otro lado, lo que ocurre después de un año o más. Puede afectar también la cara, lengua y mandíbula, pero típicamente el Parkinson no afecta solo la cabeza, como el temblor esencial. A diferencia de los temblores esenciales y cerebelosos, el temblor parkinsónico suele presentarse en reposo y atenuarse o desaparecer con el movimiento. Puede reaparecer al terminar el movimiento. El temblor postural puede preceder y coexistir con el temblor en reposo, y ambos son comunes con el envejecimiento.
La rigidez representa un aumento uniforme en la resistencia a los movimientos pasivos durante todo el rango de movimiento de los grupos musculares que actúan sobre una articulación. Puede haber interrupciones breves y regulares de la resistencia durante el movimiento pasivo, que corresponden a temblor subclínico y pueden dar la sensación de una rueda dentada. La combinación de rigidez y distonía típicamente provoca flexión del cuello, inclinación hacia adelante de la porción superior del tronco y tendencia a mantener los brazos en una posición en flexión y aducción.
La inestabilidad postural es una de las características más limitantes y refractarias del parkinsonismo y es un factor contribuyente importante para las caídas, lesiones y menor locomoción. Aunque puede observarse disminución en los reflejos posturales al principio de la EP, rara vez contribuye un problema hasta que la enfermedad progresa. De hecho, la inestabilidad postural significativa en fases tempranas del padecimiento sugiere un diagnóstico diferente a EP.
La marcha parkinsónica se caracteriza por pasos cortos, arrastrados y con tendencia a voltearse en bloque. En las fases avanzadas de la enfermedad se dificulta el iniciar, detener la marcha y voltearse. Puede ocurrir marcha festinante, en un esfuerzo aparente del paciente por alcanzar el centro de gravedad de su cuerpo. Sin embargo, la presencia de una marcha atáxica o atípica sugiere otro diagnóstico, como hidrocefalia o atrofia sistémica múltiple con afección cerebelosa. El quedarse inmóvil "congelado" es una característica de la EP avanzada y ocurre sobre todo en relación con el inicio de la marcha (duda para iniciar), al intentar cambiar de dirección o al entrar en un área muy concurrida o angosta. A diferencia de la mayoría de los signos motores parkinsónicos, esta alteración puede exacerbarse con el tratamiento dopaminomimético en dosis altas.
Condiciones asociadas
Además del parkinsonismo, muchos pacientes presentan alteraciones cognitivas, de ánimo, sensoriales, de sueño y autonómicas. Los cambios en la función cognitiva, de comportamiento y de ánimo son resultado en parte de la alteración de otros circuitos no motores de los ganglios basales. Aunque la demencia no es una característica temprana de la EP, ocurre hasta en el 30 porciento de los pacientes.19,20 La demencia parece ocurrir con más frecuencia en pacientes que tienen características principalmente aquinéticas-rígidas y suele ser precedida por alucinaciones inducidas por medicamentos y depresión. Desde el punto de vista patológico, los pacientes con demencia tienen patologías mixtas, que pueden incluir enfermedad de Alzheimer y cambios de enfermedad difusa de los cuerpos de Lewy.
La depresión afecta hasta al 50 porciento de los pacientes con EP. Son comunes los cambios de personalidad, caracterizados por apatía, ausencia de asertividad e indecisión. Puede ocurrir depresión en cualquier fase de la enfermedad, y correlaciona poco con la severidad de la misma.21-23 Debido a que el retraso psicomotor de la depresión puede ser muy semejante a las características hipoquinéticas del parkinsonismo, la depresión puede ser difícil de detectar. Esta puede ser inducida o agravada en forma iatrogénica por la polifarmacia antiparkinsónica. Por lo tanto, debe consderarse la simplificación del tratamiento antes de que el médico decida agregar farmacoterapia para la depresión.
Los trastornos del sueño son comunes en la EP. Muchos pacientes no duermen suficiente y otros tienen sueño de mala calidad. El sueño de movimientos oculares rápidos suele estar muy fragmentado. La somnolencia diurna y las siestas frecuentes son signos típicos de interrupción del sueño. Los factores que alteran el sueño incluyen reaparición del parkinsonismo durante la noche en forma de bradicinesia y rigidez (se dificulta el voltearse en la cama) y temblor o movimientos involuntarios (v.gr., mioclonías y movimientos periódicos de las piernas) que se deben a la reducción del efecto de los medicamentos antiparkinsónicos.24 Los sueños vívidos y las alucinaciones, que pueden ser efectos adversos del tratamiento antiparkinsónico, pueden contribuir también a los trastornos del sueño.25 Además de las alteraciones relacionadas con la enfermedad y los medicamentos, con frecuencia ocurren apnea del sueño y otros trastornos en los pacientes ancianos.24 Los datos sobre las características del sueño suelen brindar información útil, y el tratamiento exitoso de estos trastornos mejora en forma típica la capacidad funcional durante el día. Otras alteraciones no motoras en la EP incluyen disfunción sexual, disfunción urinaria y constipación.
Tratamiento
Los objetivos del tratamiento son mantener la función controlando la alteración primaria con tratamiento sintomático y minimizar la incapacidad secundaria (contracturas, artritis acelerada) por medio de un programa constante de ejercicio físico. Ocurrió un gran revuelo en 1969, cuando Cotzias demostró que la administración oral del precursor de dopamina levodopa podía aliviar el parkinsonismo.26 El tratamiento con levodopa se convirtió en la base del tratamiento y mejoró mucho la calidad de vida de los pacientes con EP, proporcionándoles una expectativa de vida casi normal. Sin embargo, pronto fue evidente que la levodopa no detenía la progresión de la enfermedad y, más alarmante, que el beneficio terapéutico se asociaba con complicaciones motoras y cognitivas asociadas con el fármaco.27,28
Se cree que las estrategias terapéuticas tempranas afectan en forma significativa la progresión tardía de los síntomas y que el manejo equivocado de estas estrategias puede contribuir a inducir incapacidad secundaria. Sin embargo, existe controversia sobre qué constituye el manejo farmacológico temprano óptimo de la EP.29 Los puntos de preocupación son la contribución potencial de la dopamina para acelerar la progresión de la enfermedad a través de la formación de radicales libres tóxicos y el desarrollo tardío de complicaciones motoras relacionadas con los fármacos.30,31 Sin embargo, existen pocas evidencias clínicas convincentes que apoyen estas preocupaciones. Para evaluar estos aspectos un estudio patrocinado por los Institutos Nacionales de Salud (NIH de los EUA) investiga el uso de dosis altas y bajas de carbidopa-levodopa.
Desde un punto de vista práctico, la levodopa debe iniciarse en cuanto el paciente encuentre que los síntomas interfieren con su empleo o estilo de vida. Las estrategias para ahorrar levodopa, como retrasar su inicio e iniciar agonistas de dopamina u otros agentes menos potentes en las fases tempranas de la enfermedad, pueden estar justificadas en algunos casos. Sin embargo, en general, la monoterapia con estos agentes rara vez tiene el mismo valor terapéutico que el tratamiento con levodopa. Esta opinión puede requerir reevaluación al introducirse los nuevos agonistas de dopamina de acción prolongada que se supone controlan en forma adecuada los síntomas y los nuevos monoamino oxidasa tipo B (MAO B) e inhibidores de la MAO AB.
El tratamiento sintomático de la EP debe individualizarse. Por ejemplo, los pacientes con inicio temprano representan una categoría especial. En ellos es importante descartar el parkinsonismo secundario, como la enfermedad de Wilson, la ataxia espinocerebelosa tipo 3 (enfermedad de Machado-Joseph) o la EP inducida por medicamentos [ver tabla 1]. Los autores recomiendan que estos pacientes sean referidos a un especialista en trastornos del movimiento para evaluación y tratamiento. Es importante tratar de retrasar el desarrollo de complicaciones motoras inducidas por la levodopa en los pacientes jóvenes.
La carbidopa-levodopa es la mejor elección para el tratamiento inicial de todos los pacientes con EP. La carbidopa es un inhibidor periférico decarboxilasa que bloquea la ruptura periférica de la levodopa. Por lo tanto, la carbidopa minimiza los efectos adversos asociados con la conversión periférica de levodopa a dopamina. Aunque no existen evidencias para elegir un esquema diferente a la carbidopa-levodopa en dosis bajas, el tratamiento combinado temprano (v.gr., con amantadina, anticolinérgicos o agonistas dopaminérgicos) puede estar justificado mientras controle los síntomas en forma adecuada.
Levodopa y sus fórmulas La levodopa está disponible en fórmulas de liberación inmediata regular de carbidopa más levodopa (10 mg de carbidopa/25 mg de levodopa, 25 mg/100 mg y 25 mg/250 mg) y de liberación controlada (LC) (25 mg/100 mg, 50 mg/200 mg). Los suplementos de carbidopa (25 mg al día a 200 mg al día) se usan en ocasiones para tratar a algunos pacientes con náusea persistente, al parecer en respuesta a dosis inadecuadas de carbidopa. Para pacientes con náusea persistente el antiemético débil trimetobenzamida (para pacientes externos) y el ondansetrón intravenoso u oral (para hospitalizados) son bien tolerados y eficaces. Otros antieméticos, como los antagonistas del receptor de dopamina, proclorperacina y metoclopramida, deben evitarse porque pueden empeorar en forma importante los síntomas parkinsónicos.
El tratamiento con carbidopa-levodopa de LC permite la comodidad de menos dosis durante el día y una respuesta clínica más gradual en pacientes con fluctuaciones motoras leves a moderadas (supresión).32 En un estudio doble ciego de cinco años de duración que comparó la fórmula carbidopa-levodopa de liberación inmediata regular con la controlada, la segunda fue más eficaz para disminuir la progresión de la incapacidad, medida por varios indicadores de calidad de vida. Sin embargo, sorprendentemente, pareció que la carbidopa-levodopa LC no tuvo ventajas sobre la regular para disminuir la incidencia de fluctuaciones motoras.33 Solo el 16 porciento de los pacientes en ambos grupos desarrollaron fluctuaciones motoras, un porcentaje muy bajo comparado con reportes más tempranos. Esta baja incidencia de discinesias puede deberse en parte con las dosis relativamente bajas de levodopa que se han usado (200 a 400 mg/día).
El inicio del tratamiento con las fórmulas de uso regular o de LC de carbidopa-levodopa es aceptable. El precio de la forma regular, que en la actualidad está disponible en su fórmula genérica, es menor que el de la fórmula de LC. Aunque la carbidopa-levodopa regular se absorbe mejor con el estómago vacío, suele administrarse con los alimentos para disminuir la náusea. Por el contrario, la absorción y biodisponibilidad de las fórmulas de LC aumenta cuando se toma con alimentos.32 Los efectos adversos de la carbidopa-levodpa pueden controlarse iniciando con una dosis pequeña (v.gr., media tableta de carbidopa-levodopa, 25 mg/100 mg bid) y después aumentando la dosis según se tolere hasta que se presente náusea. El ajuste de la dosis debe hacerse despacio porque la respuesta total a cada incremento se presenta en varias semanas. En la mayoría de los casos la dosis de levodopa no debe exceder 300 a 400 mg/día en las fases tempranas de la enfermedad. Pueden requerirse dosis más altas y más frecuentes en pacientes con enfermedad más avanzada que presentan fluctuaciones motoras.
Si se selecciona la carbidopa-levodopa LC como tratamiento inicial, la dosis es la mitad de una tableta de 50 mg/200 mg dos veces al día. Los autores prefieren que el paciente inicie tomando el medicamento en el desayuno y la comida, con un intervalo de cinco a seis horas en lugar del esquema estricto de nueve a 12 horas. El objetivo es obtener una meseta en la concentración plasmática de levodopa en la parte inicial del día y dejar que esta meseta se reduzca durante el resto del día. Eventualmente el paciente requerirá dosis en la tarde y noche. Debido a la menor biodisponibilidad de la levodopa en la fórmula de LC (77 porciento en comparación con la fórmula regular), la fórmula de LC de 600 mg equivale a alrededor de 400 mg de la fórmula de liberación inmediata. Por último, se ha reportado que la fórmula de LC induce o agrava los síntomas distónicos, comparado con la carbidopa-levodopa regular, en especial en pacientes con inicio temprano.32
Estrategias para aumentar la levodopa Otras estrategias para aumentar la trasmisión de dopamina incluyen el bloqueo metabólico de la dopamina por inhibición de la enzima MAO B.34 La seliginina es un inhibidor selectivo y reversible de la MAO B que tiene un efecto sintomático débil cuando se usa solo y un efecto sintomático modesto cuando se usa con carbidopa-levodopa. La seliginina se emplea para aliviar la supresión leve o tumor residual y evitar aumentar la dosis de levodopa. Está disponible en cápsulas o tabletas de 5 mg. La dosis habitual es de 5 mg con el desayuno y la comida. Mientras se mantenga esta dosis, no se requieren restricciones dietéticas paa evitar las crisis hipertensivas inducidas por tiramina, a diferencia de lo que sucede con los inhibidores no selectivos (MAO AB) y los inhibidores de la MAO A, para los que se requiere restricción dietética. El insomnio es un efecto adverso significativo de la seliginina, en especial cuando se toma después de mediodía. Las personas mayores y las que tienen enfermedad cardiaca estable pero importante pueden beneficiarse de dosis tan bajas como 2.5 mg/día. La dosis debe disminuirse o eliminarse si ocurren efectos adversos hiperdopaminérgicos refractarios (v.gr., empeoramiento de la discinesia, alucinosis o confusión).
Una estrategia novedosa puede aumentar la duración de la respuesta a la levodopa y aliviar las fluctuaciones motoras.35 La O-metilación de la levodopa a 3-O-metildopa por la catecol-O-metiltransferasa (COMT) hace que la levodopa esté menos disponible para conversión a dopamina y por tanto sea menos eficaz. Debido a que se encuentra actividad de COMT en el tejido periférico y el cerebro, los inhibidores de la COMT aumentan la absorción de la levodopa y disminuyen su eliminación. El retraso en la eliminación de la levodopa puede reducir las fluctuaciones motoras clínicas y los requerimientos de levodopa. El entacapone y el tolcapone son dos inhibidores de la COMT que se evalúan en la actualidad en estudios de fase III.
Agonistas de dopamina Los agonistas de dopamina no requieren transformación o transporte facilitado a través de la barrera hematoencefálica, y actúan directamente sobre los receptores posinápticos de dopamina. Dos de éstos, la bromocriptina y el pergolide, actúan mejor como adyuvantes del tratamiento con levodopa y no son tan eficaces como ésta última. Sus efectos adversos (confusión, alucinaciones, trastornos del sueño) y la relación riesgo-beneficio son menos favorables que los de la carbidopa-levodopa. Cuando los agonistas de dopamina se usan como monoterapia en tratamientos ahorradores de levodopa, la mayoría de los pacientes requieren de la adición de levodopa después de seis a 18 meses para mantener un control adecuado de los síntomas. Además, un estudio extenso no pudo demostrar ningún beneficio a largo plazo de la monoterapia temprana con agonistas.36 Dos agonistas nuevos, el pramipexol y el ropinirol, pueden ser mejores agentes para monoterapia y tener acción más prolongada. La dosis inicial del pramipexol es de 0.125 mg tres veces al día. Al final de la primera semana la dosis puede aumentarse a 0.25 mg tres veces al día, con incrementos semanales de 0.25 mg tres veces al día, hasta una dosis máxima de 1.5 mg tres veces al día, con o sin levodopa. La dosis inicial del ropinirol es de 0.25 mg tres veces al día. Dependiendo de la respuesta, la dosis puede aumentarse en forma semanal 0.25 mg tres veces al día hasta 1 mg tres veces al día. Después de la semana 4 la dosis diaria puede aumentarse a 1.5 mg/día en forma semanal, hasta una dosis máxima de 9 mg/día, y después aumentarse 3 mg/día hasta una dosis máxima de 24 mg/día. El uso de los agonistas de dopamina en las fases tardías de la enfermedad se justifica en casos seleccionados porque estos agentes pueden tener fluctuaciones motoras suaves y pueden disminuir las discinesias. Sin embargo, esto requiere de un ajuste cuidadoso de la dosis y de ajustes simultáneos en el resto del tratamiento antiparkinsónico.
Amantandina y anticolinérgicos Algunos médicos prefieren tratar la EP leve con agentes menos potentes, como la amantadina y anticolinérgicos, para retrasar el inicio de la levodopa, incluso a pesar de que no existen datos de que esta maniobra evite la aparición de las discinesias. Sin embargo, es lógico iniciar el tratamiento con estos agentes menos potentes en los pacientes jóvenes con EP, en especial en los que tienen distonía importante. Los pacientes con temblor predominante pueden también responder en forma satisfactoria a la amantadina y a los anticolinérgicos.
El efecto protector potencial de la amantadina puede ser mediado por su débil efecto antagonista del glutamato. En la actualidad se evalúan antagonistas del glutamato más potentes y selectivos para determinar su eficacia como adyuvantes en el tratamiento antiparkinsónico.37 Un estudio clinicopatológico reciente sugiere que la amantadina puede tener también un efecto neuroprotector sobre la progresión de los síntomas y la muerte de las células del área negra.38 Por último, se ha sugerido que la amantadina es un adyuvante útil a la levodopa en fases más avanzadas de EP y puede reducir la severidad de las discinesias inducidas por medicamentos. Los efectos adversos de la amantadina son edema, eritema y livedo reticularis. En los pacientes ancianos puede agravar la confusión y la psicosis. La dosis inicial es de una tableta de 100 mg dos veces al día; en caso necesario la dosis puede aumentarse a tres veces al día después de una semana. Debido a que la amantadina se elimina por los riñones, puede acumularse con rapidez hasta niveles tóxicos en pacientes con daño renal.
Los anticolinérgicos se han usado por mucho tiempo en la EP, en especial por sus efectos sobre el temblor, la rigidez y la distonía. Sus efectos sobre la acinesia, la bradicinesia, la postura y la marcha son limitados. Las principales desventajas, en especial en los ancianos, son el deterioro de la memoria, las alucinaciones, la visión borrosa, la constipación, la polaquiuria y la xerostomía. Este último efecto adverso puede ser útil en los pacientes con sialorrea importante, que se asocia con frecuencia con la EP.
Tratamiento neuroprotector Los mecanismos de la muerte de las células del área negra parecen incluir el estrés por oxidantes, que suele agravarse por el alto contenido de los compuestos reactivos, dopamina y melanina, en estas células. La selegilina tiene propiedades antioxidantes in vitro. En estudios experimentales en animales, la seligilina puede bloquear la neurotoxicidad dopaminérgica de la 1-metil-4-fenil-1,2,3,6-tetrahidropiridina (MPTP) al bloquear en forma selectiva la formación del metabolito de la MPTP, el ión 1-metil-4-fenilpiridinio (MPP+), una toxina con alto poder oxidativo para las neuronas de dopamina.17,39 Debido a que el MPTP también produce un síndrome semejante a la EP en humanos, se ha postulado que la selegilina podría reducir las fuentes intrínsecas y extrínsecas de estrés oxidante en las neuronas dopaminérgicas humanas. Su uso en la EP como agente neuroprotector fue apoyado por los reportes del grupo DATATOP (Tratamiento del parkinson con tratamiento antioxidante con tocoferol y deprenil), que demostraron que la monoterapia con selegilina retrasaba en forma significativa la progresión de los síntomas durante un año.39,40 La interpretación de estos resultados se confundió con un efecto sintomático leve e inesperado de la selegilina que pudo haber causado el retraso de la progresión de los síntomas cuando se comparó con el grupo tratado con placebo.17 El seguimiento y el análisis posterior de los datos del DATATOP y de varios estudios pequeños sobre selegilina no han revelado ninguna reducción sustancial inducida por selegilina en la progresión de los síntomas después de un año.41-43 Por lo tanto, los datos clínicos disponibles no apoyan el concepto de que la selegilina pueda tener un efecto benéfico sobre la EP a largo plazo. Un solo estudio europeo reportó mayor mortalidad con la seleginina, pero este dato no concuerda con los estudios previos, por lo que es poco confiable.44
Tratamiento de la enfermedad de Parkinson avanzada
El manejo clínico de la EP avanzada se vuelve cada vez más complejo al empeorar la marcha y existir inestabilidad postural, con aumento de los periodos estáticos, las caídas y la aparición de fluctuaciones motoras y discinesias. Igualmente incapacitantes son los periodos confusionales inducidos por medicamentos, las alucinaciones y la psicosis. En esta fase está indicado referir al paciente con un especialista en trastornos del movimiento. Lo siguiente es solo una breve descripción de los aspectos más importantes en el manejo de la EP avanzada.
Supresión Típicamente, la supresión de la acción de la levodopa es el signo más temprano de fluctuaciones motoras.27 La administración de medicamentos antiparkinsónicos antes de los alimentos en lugar de con ellos puede evitar la absorción errática y parcial que contribuye a este problema. La carbidopa-levodopa regular puede combinarse con la fórmula de liberación controlada (v.gr., media tableta de liberación inmediata de carbidopa-levodopa 10/100 mg y una tableta de LC de 50/200 mg) para proporcionar un mejor balance entre las respuestas inmediata y sostenida. Los agonistas de la dopamina y la selegilina tienen una vida media más larga que la carbidopa-levodopa, y producen una respuesta clínica más suave que la primera usada en forma aislada en los pacientes que presentan fluctuaciones motoras. Otra estrategia consiste en acortar los intervalos de las dosis. Sin embargo, los esquemas con dosis más frecuentes que cada cuatro horas son difíciles de manejar desde el punto de vista clínico y probablemente deben usarse solo en clínicas especializadas de trastornos del movimiento.
Fenómeno presente-ausente Ocurren cambios súbitos e impredescibles en las respuestas antiparkinsónicas mezcladas con discinesias, descargas autonómicas (diaforesis profusa, urgencia urinaria) y cambios en el estado de ánimo en algunos pacientes con EP avanzada, a lo que se le llama fenómeno de presente-ausente (on-off).45 Las estrategias para controlar la supresión (ver antes) suelen ser de valor muy limitado en la mayoría de los pacientes que presentan este fenómeno. Se requieren estrategias más agresivas para estabilizar la variabilidad en la concentración en plasma de la dopamina y por lo tanto la respuesta clínica. Estas estrategias incluyen dietas de redistribución de proteínas y, por lo tanto, respuesta clínica. Las dietas consisten en mantener la ingesta proteína y calórica óptima diaria cambiando el consumo de proteínas principalmente a las comidas de la tarde. Esto minimiza la competencia diurna entre la levodopa y otros aminoácidos neutros grandes por el transporte a través de la barrera hematoencefálica. Los pacientes que sufren del fenómeno presente-ausente son también candidatos ideales para la neurocirugía estereotáctica.
Cambios en el comportamiento inducidos por drogas Los pacientes con EP pueden sufrir cambios en la personalidad inducidos por los medicamentos que son lentos pero profundos, y que se expresan como actitudes temperamentales, ilógicas y demandantes, egocentrismo y aparente desinterés por las necesidades de otros. Esto puede ser un preludio de una depresión agitada o psicosis inducida por los medicamentos o secundaria a la aparición de demencia. La psicosis inducida por drogas se caracteriza por alucinaciones visuales con retención del yo interno. las alucinaciones auditivas sugieren depresión psicótica coexistente o demencia, pero pueden ser un efecto adverso de los medicamentos anticolinérgicos.47 En muchos casos los síntomas cognitivos y psiquiátricos cesan con la eliminación de los anticolinérgicos y la amantadina. Sin embargo, en algunos pacientes es necesario disminuir las dosis de dopaminomiméticos o suspenderlos, típicamente en el siguiente orden: selegilina, dosis noctura de antagonista de dopamina o carbidopa-levodopa de LC y, por último, carbidopa-levodopa regular. Si el paciente mejora después de un ajuste en la polifarmacia, el impacto sobre los síntomas parkinsónicos motores irá de negligible a positivo. Si el paciente requiere una reducción drástica en el tratamiento antiparkinsónico, es probable que el empeoramiento de los síntomas motores sea intolerable. No se recomiendan ya los días aislados sin levodopa porque varios reportes indican que son potencialmente peligrosos y que no proporcionan un beneficio significativo a largo plazo.
Cuando la reducción en la polifarmacia antiparkinsónica es intolerable, puede requerirse un medicamento antipsicótico, y la clozapina es el fármaco de elección en la mayoría de los pacientes. La clozapina es un antipsicótico atípico muy eficaz, con propiedades bloqueadoras de los receptores dopaminérgico (D4) y serotoninérgico (S2) y pocos efectos adversos extrapiramidales. La clozapina puede tener efectos adicionales antitemblor y antidiscinéticos. Sus efectos adversos más persistentes son hipotensión ortostática y sialorrea. En los pacientes con demencia puede causar confusión temporal hasta por una semana. La clozapina se asocia con un riesgo de uno porciento de agranulocitosis y requiere de vigilancia semanal de la cuenta de leucocitos. La dosis debe ajustarse con cuidado entre 12.5 y 100 mg/día. Las dosis mayores de 150 mg/día pueden agravar los síntomas motores del Parkinson. Otros antipsicóticos atípicos que no se asocian con agranulocitosis incluyen el risperidone (0.5 a 2 mg/día) y el olanzapine (5 a 15 mg/día). Según la experiencia de los autores, el risperidone tiende a agravar los síntomas parkinsónicos con el tiempo. Por el contrario, un estudio abierto de olanzapina (5 a 15 mg/día) sugiere que este medicamento puede aliviar la psicosis inducida por medicamentos sin agravar los síntomas del Parkinson. Sin embargo, experiencias no publicadas de la institución de los autores que incluyeron a 20 pacientes sugieren que este medicamento puede no ser tan bien tolerado como la clozapina. Otros antipsicóticos alternativos que son tolerados ocasionalmente por pacientes individuales incluyen los neurolépticos de baja potencia, como la mesoridacina (10 mg/día) y el molindone (5 a 15 mg/día).
Depresión Los pacientes con depresión típicamente responden a los antidepresivos (v.gr., tricíclicos e inhibidores selectivos de la recaptura de serotonina [ISRS]). Los autores prefieren prescribir ISRS de acción corta más que de acción prolongada, ya que existen reportes de que la fluoxetina, un ISRS de acción prolongada, puede agravar la EP. Además, la combinación de fluoxetina y selegilina conlleva el riesgo teórico de un síndrome hiperserotoninérgico (delirio con mioclonía e hiperpirexia). Sin embargo, en la experiencia de los autores, no han observado ni una sola reacción hiperserotoninérgica en más de 30 pacientes tratados con esta combinación. Por último, se recomienda el tratamiento electroconvulsivo (TEC) para los pacientes con EP que sufren de depresión refractaria o que no toleran los antidepresivos orales. Existen varios reportes que indican que el TEC puede ser benéfico también para los síntomas parkinsónicos motores.
Tratamientos neuroquirúrgicos Existe nuevamente interés en el tratamiento quirúrgico del parkinsonismo intratable. En la actualidad se prefiere la palidotomía, una operación realizada por vez primera en los años 50 y después abandonada en favor de la talamotomía. Estudios clínicos recientes sobre la palidotomía han demostrado un beneficio claro en las características cardinales de la EP (acinesia/bradicinesia, temblor y rigidez), así como reducción marcada en las discinesias inducidas por medicamentos y las fluctuaciones motoras.5,50-52 Es importante no retrasar la cirugía demasiado porque el costo, en términos de discapacidad social, pérdida de independencia y autoconfianza, pérdida del empleo y deterioro en la calidad de vida, es grande y no siempre reversible. Los pacientes más jóvenes con discinesia inducida por medicamentos e intratable son los candidatos ideales. Otros tipos de tratamiento quirúrgico están actualmente en estudio (v.gr., estimulación cerebral profunda y trasplante mesencefálico fetal y de líneas celulares tratadas por ingeniería genética), y quizá demuestren ser seguros y eficaces. Los pacientes deben ser referidos a un especialista en trastornos del movimiento para ser evaluados antes de la cirugía. La presencia de demencia o atrofia cerebral importantes constituyen contraindicaciones. Además, los pacientes con formas de parkinsonismo diferentes a la EP no responden en forma favorable a la palidotomía.
OTRAS FORMAS DE PARKINSONISMO
Debido a que la tasa de errores diagnósticos en los casos de EP confirmados por autopsia es hasta del 25 porciento, es claro que otras formas de parkinsonismo pueden parecerse mucho a la EP.18 Además de satisfacer los criterios para diagnosticar la EP, es importante poner en duda el diagnóstico en las siguientes circunstancias: cuando existe historia de exposición reciente a medicamentos o toxinas que puedan causar parkinsonismo, cuando existe historia de traumatismo cerebral repetido, si los síntomas tienen inicio bilateral, si existe demencia temprana, cuando hay signos de inestabilidad postural prominente, disfunción autonómica o signos cerebelosos o piramidales. Los trastornos que se analizan a continuación son los que más se confunden con EP.
Parkinsonismo inducido por medicamentos
El parkinsonismo inducido por medicamentos es muy semejante a la EP excepto por el temblor, que suele ser (aunque no siempre) menos importante. Con frecuencia es causado por el uso subagudo o crónico de neurolépticos, carbonato de litio, antieméticos o bloqueadores de los canales del calcio.53 En una publicación europea reciente, alrededor del 30 porciento de los pacientes parkinsónicos en una clínica de trastornos del movimiento tuvieron parkinsonismo inducido por medicamentos.54 Los síntomas parkinsónicos casi siempre desaparecen días o semanas después de suspender el agente agresor. Las mujeres ancianas son especialmente propensas a sufrir este síndrome, y hasta el 40 porciento permanece afectada después de suspender el medicamento, lo que sugiere que el medicamento desenmascaró en forma prematura un fenotipo parkinsónico. Los agentes relacionados con más frecuencia han sido los bloqueadores de los canales del calcio (diltiacem y verapamil) y los antagonistas de dopamina (metoclopramida y neurolépticos).55 Sin embargo, en la experiencia de los autores, los bloqueadores de los canales del calcio causan el síndrome con mucho menos frecuencia que los antagonistas de la dopamina. Son causas menos comunes de parkinsonismo inducido por fármacos el ácido valproico y, recientemente, la fluoxetina. El parkinsonismo puede ser inducido también por la administración prolongada de agentes antihipertensivos como la reserpina, que depleta las reservas presinápticas de dopamina, y la metildopa, que bloquea la producción de dopamina. El parkinsonismo leve inducido por medicamentos puede ser aliviado por agentes anticolinérgicos, amantadina y levodopa. Los adultos jóvenes que presentan un síndrome parkinsónico deben ser interrogados respecto al uso de drogas ilegales, en espcial narcóticos sin marca que pueden ser MPTP. La exposición a sustancias tóxicas también puede causar un estado parkinsónico [ver tabla 1].
Parálisis supranuclear progresiva
La parálisis supranuclear progresiva (PSP) es uno de los síndromes parkinsónicos más comunes. Es un padecimiento diferente de etiología desconocida y que comienza típicamente en la sexta o la séptima décadas de la vida y progresa a la muerte en cinco a 10 años.55
Características clínicas Al inicio de la enfermedad son comunes las molestias sobre deterioro visual, mareo, inestabilidad, respuestas lentas, caídas y deterioro del lenguaje. El temblor es muy poco común. La expresión facial, con los ojos muy abiertos, el ceño fruncido y la cabeza hiperextendida proporciona una imagen inolvidable que contrasta con la fascies de máscara y la postura en flexión de la EP [ver figura 4]. La parálisis supranuclear característica que afecta los movimientos de los ojos hacia abajo suele ser el primer signo, seguido de deterioro de los movimientos oculares horizontales. Sin embargo, en algunos casos las características parkinsónicas son prominentes y no existen los datos oculomotores característicos, lo que puede causar un diagnóstico erróneo. El resto del examen suele revelar rigidez axial prominente (en especial de la nuca), distonía proximal y signos de motoneurona superior y cerebelosos. Un número significativo de personas con PSP puede desarrollar demencia. Se presenta degeneración diseminada y de grado variable en el tallo cerebral, los ganglios basales y el cerebelo. Aunque puede ocurrir cierta respuesta con los medicamentos antiparkinsónicos, en especial en las fases tempranas, el tratamiento no es muy eficaz.

|
| Figura 4 |
| Expresión de un paciente con parálisis supranuclear progresiva. |
Atrofia sistémica múltiple
Las atrofias sistémicas múltiples (ASM) son un grupo clínicamente diverso de trastornos neurodegenerativos caracterizados por diversos grados de parkinsonismo, disfunción cerebelosa y falla autonómica [ver figura 5a y b]. La edad promedio de inicio de la enfermedad son 50 años y la supervivencia promedio es de seis años.
|
|
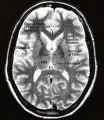
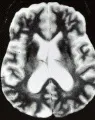
|
|
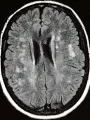
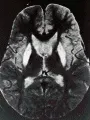
Dependiendo de las características clínicas que predominen, estos padecimientos se han agrupado en tres categorías amplias: degeneración nigroestriada, atrofia olivopontocerebelosa (AOPC) y síndrome de Shy-Drager. Los pacientes con parkinsonismo relativamente puro se clasifican como con degeneración nigroestriada. Los que tienen una combinación de parkinsonismo y otros datos, como ataxia, afección de neurona motora superior y corticobulbar, mioclonía, alteraciones oculomotoras, neuropatía periférica y sordera, se agrupan en la AOPC. Las AOPC son un grupo muy heterogéneo que incluye formas de la enfermedad tanto hereditarias como esporádicas. Solo la forma esporádica de la AOPC es parte del espectro de ASM. Si las características de parkinsonismo se asocian con signos prominentes de falla autonómica, se justifica el diagnóstico de síndrome de Shy-Drager.
Típicamente se encuentra degeneración diseminada en los ganglios basales, la médula espinal, el tallo cerebral y el cerebelo. Una característica patológica única y distintiva de la ASM es la presencia de inclusiones citoplásmicas gliales. Aún no es claro el papel de éstas en el proceso patológico.
Tratamiento En general, las características parkinsónicas de la ASM tienen una respuesta solo modesta a los medicamentos antiparkinsónicos, aunque la respuesta suele ser mejor que en la parálisis supranuclear progresiva. El tratamiento dopaminomimético en estos padecimientos suele estar limitado por la hipotensión ortostática. Ocurren discinesias inducidas por medicamentos en la ASM pero, a diferencia de la EP, suelen afectar más la cara y el cuello.
El tratamiento de la hipotensión ortostática en los pacientes con ASM (así como EP y otros trastornos asociados con hipotensión ortostática sintomática) incluye medidas simples como cambios de posición lentos, aumentar la ingesta de sal y líquidos y evitar las habitaciones demasiado calientes, las comidas abundantes y el alcohol. Otras medidas incluyen el empleo de medias elásticas, dormir en posición de Trendelenburg y ajustar otros tratamientos farmacológicos que puedan afectar la presión arterial. Debido a que la presión arterial tiende a ser mayor en la tarde, los pacientes pueden aprender a realizar la mayor parte de sus actividades cotidianas a esa hora. Si estas medidas fallan, se han propuesto diversas medida farmacológicas para tratar la insuficiencia autonómica.
Otros síntomas, como la impotencia, la urgencia urinaria y la incontinencia, así como las tormentas autonómicas con diaforesis profunsa, son más difíciles y en ocasiones imposibles de tratar por medio de fármacos. En ocasiones la impotencia mejora con la yohimbina. Los implantes de pene pueden ayudar, pero no deben realizarse sino hasta que los cambios ortostáticos estén bien controlados. La constipación puede manejarse con fibra en dosis altas, hidratación, bisacodil, concentrado de sena o lactulosa. La urgencia urinaria y polaquiuria pueden responder a la oxibutinina y al sulfato de hiosciamina; la incontinencia requiere de sondeo intermitente, uso de pañales o ambos. La gastroparesia puede mejorar con el uso de 5 a 10 mg de cisaprima antes de los alimentos; las dosis mayores pueden agravar el temblor parkinsónico.
Degeneración cortical de los ganglios basales
La degeneración cortical de los ganglios basales (DCGB) ocurre casi con tanta frecuencia como la parálisis supranuclear progresiva. El síndrome comienza típicamente en la sexta década de la vida y se manifiesta como un síndrome variado pero característicamente unilateral con rigidez, distonía, movimientos lentos y apraxia, con o sin pérdida sensorial cortical.57 Los síntomas empeoran hasta que el paciente se vuelve totalmente incapacitado. Las imágenes de resonancia magnética y patología macroscópica revelan pérdida cortical focal prominente en las áreas parietal y frontal contralateral. El examen histológico revela gliosis y neuronas edematizadas (como globos) acrómáticas con pérdida neuronal. Existen también alteraciones en la sustancia blanca que consisten en edema y desmielinización axonal, y el neuropilo tiene una apariencia espongiforme. Además de afectar la corteza, estos cambios alteran el núcleo subtalámico, el globo pálido, el nucleo estriado, el tálamo y varios nucleos cerebelosos. Los tratamientos suelen ser ineficaces.
Parkinsonismo vascular
El concepto de parkinsonismo vascular o ateroesclerótico ha sido tema de incertidumbre y controversia durante décadas. Aunque existe un acuerdo general de que esta entidad existe, su naturaleza precisa no es clara. Por lo general, los pacientes con parkinsonismo vascular muestran un síndrome aquinético-rígido sin temblor. Muchos tienen marcha de pequeños pasos.58 Además, la mayoría tiene historia de insuficiencia cerebrovascular y datos neurológicos asociados, como signos de neurona motora superior, parálisis seudobulbar y demencia, que los distingue de los pacientes con EP. Los estudios de imagen son heterogéneos y pueden revlear lagunas en los ganglios basales o infartos múltiples [ver figura 5c]. La microangiopatía hipertensiva o diabética y la enfermedad de Binswanger típicamente afecta la sustancia blanca en forma difusa o en parches confluentes en el centro semioval.58 Otras enfermedades que presentan un cuadro semejante en forma ocasional son las vasculitis inflamatorias, el síndrome antifosfolípico y, en raros casos, leucoencefalopatías como la leucodistrofia hereditaria de inicio en el adulto. Se requiere un examen de necropsia para realizar el diagnóstico definitivo debido a la ausencia de marcadores claros de la enfermedad. Desde el punto de vista terapéutico, la respuesta a los agentes dopaminérgicos es limitada, y los anticolinérgicos y la amantadina con frecuencia causan confusión y delirio.
Parkinsonismo y demencia
Existen diversas enfermedades (v.gr., enfermedad de Alzheimer [EA], de Creuzfeld-Jakob y de Pick) que tienen como datos principales tanto demencia como signos de afección de los ganglios basales. Por ejemplo, en el caso de la EA, el 25 porciento de los pacientes tienen rigidez leve y bradicinesia. Hasta el 40 porciento de los casos de EP tienen cierta patología de EA.59 Estos pacientes típicamente presentan una forma rígida aquinética de parkinsonismo, sin temblor. El curso de la enfermedad es más rápido y el tratamiento es más difícil por la alta incidencia de efectos adversos cognitivos con el tratamiento antiparkinsónico, en especial anticolinérgicos y amantadina. La toxicidad dopaminomimética central pueden presentarse de diversas formas, incluyendo trastornos del sueño (con somnolencia diurna), cambios de personalidad, depresión y torpeza mental, confusión episódica, alucinaciones y trastornos del comportamiento.
La enfermedad difusa por cuerpos de Lewy es una forma de demencia con características parkinsónicas prominentes que cada vez se detecta con mayor frecuencia.60 En algunas familias con esta enfermedad se encuentran pacientes con EP pura por enfermedad difusa por cuerpos de Lewy. Desde el punto de vista patológico, se encuentran cuerpos de Lewy en la corteza y el tallo cerebral. Algunos pacientes muestran también patología de EA, lo que dificulta la distinción de los pacientes que manifiestan un síndrome de sobreposición EA-EP. Desde el punto de vista clínico, el temblor de intención puede preceder otras características parkinsónicas, y la demencia suele ser precedida por sedación inducida por la levodopa, mioclonías y alucinaciones. Al inicio la respuesta a la levodopa puede ser importante, lo que dificulta la distinción entre enfermedad difusa por cuerpos de Lewy y EP. La progresión de los síntomas en el primer caso parece ser intermedia cuando se compaa con la progresión en la EP y los subgrupos EP-EA.
Trastornos hiperquinéticos
Los trastornos de movimiento hiperquinético incluyen diversos movimientos involuntarios (discinesias) que pueden ocurrir aislados o en combinación, e incluso en el caso de parkinsonismo. Las hiperquinesias tienen un amplio espectro de gravedad, variando de apenas detectables (v.gr., inquietud o movimiento continuo de los dedos) a violentas (v.gr., hemibalismo), e incluso a complejas y asociadas con carga emocional (v.gr., tics focales y coprolalia en el síndrome de Tourette). Cuando las discinesias ocurren en combinación, es importante determinar cuál predomina porque esto ayudará a guiar el enfoque diagnóstico y sugerir posibles tratamientos.
La corea es quizá la forma más común de discinesia.61 El término corea, que significa danza, se refiere a movimientos arrítmicos e involuntarios, que típicamente son súbitos y breves y parecen fluir de una parte del cuerpo a otra. Cuando se combinan con movimientos más lentos, como de escritura o posiciones distónicas, la discinesia se denomina coreatetosis. El hemibalismo es un tipo de corea unilateral, de gran amplitud y violento que afecta las extremidades proximales más que las distales. La causa más común de hemibalismo es una lesión vascular del núcleo subtalámico.
Son ejemplos de otros movimientos rápidos, involuntarios que pueden confundirse con corea las mioclonías, los tics y los temblores distónicos. Las mioclonías son movimientos rápidos sin la actividad rítmica que se observa en la corea. Aunque los pacientes con mioclonías con frecuencia pierden el control motor y tiran objetos, esto rara vez sucede en los pacientes con corea o tics. A diferencia de la corea y las mioclonías, los tics motores pueden suprimirse.
Los movimientos distónicos tienden a ser más lentos y las contracciones más sostenidas (segundos o más) en comparación con la corea y los tics. Al principio la distonía puede existir solo con movimientos voluntarios (distonía de acción), pero después se presenta en forma espontánea. Las contracciones dinámicas con frecuencia causan alteraciones posturales como las de la tortícolis espasmódica. Al principio los movimientos distónicos pueden responder a trucos sensoriales, como levantar un poco la barbilla para evitar que se mueva el cuello. El temblor asociado con distonía tiende a tener una calidad brusca e irregular diferente a la calidad oscilatoria de la mayoría de los otros temblores. La corea, las mioclonías, los temblores y las contracciones distónicas son percibidas por los pacientes como involuntarias. Por el contrario, los tics con frecuencia son respuestas voluntarias a neesidades subyacentes.
COREA
Etiología
La corea se ha descrito en por lo menos 150 enfermedades somáticas como una reacción a numerosos medicamentos y toxinas.61 Desde el punto de vista médico, las causas más importantes de corea son respuestas secundarias a infecciones o procesos inmunológicos, vasculares, metabólicos, endócrinos o inducidos por fármacos [ver tablas 2 y 3]. Las enfermedades sistémicas que causan corea pueden asociarse con trastornos inflamatorios, como lupus eritematoso generalizado y poliarteritis nodosa, o con infecciones o procesos parainfecciosos como corea estreptocócica, difteria, tosferina y, más recientemente, toxoplasmosis y síndrome de inmunodeficiencia adquirida (SIDA). Los trastornos cerebrovasculares que causan corea se asocian con signos neurológicos asimétricos y un curso escalonado que es compatible con un estado lacunar.
|
||
|
|
|||
* El parkinsonismo, el temblor y la distonía inducidos por medicamentos se analizan en el texto. |
Las entidades pediátricas más importantes asociadas con corea son la corea de Sydenham, la enfermedad de Wilson, la distonía por torsión, el síndrome de Tourette y, en casos raros, la enfermedad de Huntington. Los raros casos de alteración en el metabolismo intermedio pueden causar corea, pero sus datos asociados, como encefalopatía, retraso mental y mioclonía, tienden a ser más marcados que la corea [ver tabla 4]. Los trastornos asociados con corea que pueden presentarse también en la edad adulta son la enfermedad de Wilson, la enfermedad de Huntington y la calcificación familiar de los ganglios basales (enfermedad de Fahr).
|
|||
|
Tratamiento
Independientemente de su etiología, la corea puede tratarse con depletores de dopamina (reserpina) y bloqueadores de la misma. Se prefieren los depletores de dopamina porque tienden a producir menos efectos adversos extrapiramidales que los bloqueadores y no se asocian con el riesgo de discinesia tardía. Los efectos adversos potenciales de la reserpina incluyen parkinsonismo, distonía aguda e hipotensión postural. La reserpina inicia típicamente en dosis bajas (0.10 a 0.25 mg/día) y se aumenta según se tolere (por lo general por semana) hasta que se controla la corea o aparecen efectos adversos.
COREA DE SYDENHAM
Etiología
Conocida originalmente como mal de San Vito o corea reumática, este trastorno de la infancia y rara vez de la adolescencia, se caracteriza por corea suave, fluída y sutil asociada con hipotonía, debilidad muscular y labilidad emocional. Constituye uno de los criterios mayores de Jones para fiebre reumática y afecta a las niñas con el doble de frecuencia que a los niños, pudiendo presentarse hasta seis meses después de la infección desencadenante, con duración de entre tres a seis semanas. Es una de las secuelas posinfecciosas del estreptococo §-hemolítico. A diferencia de la corea de Sydenham, otros procesos posinfecciosos asociados con corea [ver tabla 2] típicamente se presentan como encefalopatía después de los movimientos anormales.
Manifestaciones clínicas
La corea de Sydenham típicamente no se asocia con otros datos del síndrome posestreptocócico, como carditis, poliartritis, eritema marginado y nódulos subcutáneos. La corea es antecedida por fatiga, irritabilidad, inquietud, torpeza y debilidad. Son comunes las alteraciones del comportamiento y algunos pacientes desarrollan flojera mental, depresión y ansiedad. La corea recurre varias veces durante el curso de meses en el 35 porciento de los casos. En pocas ocasiones los síntomas pueden durar varios años o incluso toda la vida. En forma periódica la corea puede reaparecer, por ejemplo durante el embarazo o después del inicio de tratamiento estrogénico.
Datos de laboratorio
Debe pensarse en la posibilidad de corea de Sydenham en cualquier niños con corea, exista o no historia de faringitis previas. El diagnóstico es apoyado por pruebas de laboratorio que indican elevación en los niveles plasmáticos de antiestreptolisina O. También pueden aumentar los niveles plasmáticos de anticuerpos menos específicos, incluyendo antiestreptocinasa, antihialuronidasa, antiestreptozima y anti-ADNasa B. Los títulos de anticuerpos antineuronales pueden estar elevados en el líquido cefalorraquídeo. En algunos casos se ha asociado la presencia de anticuerpos antineuronales circulantes con sintomatología obsesivo-compulsiva. A diferencia de los pacientes con corea vascular inflamatoria secundaria a lupus eritematoso generalizado o síndrome antifosfolípido [ver tabla 2], los pacientes con corea de Sydenham no desarrollan anticuerpos anticardiolipina.
Complicaciones
Ocurren complicaciones cardiacas agudas en alrededor del 20 porciento de los pacientes, pero pueden no ser evidentes durante la evaluación inicial. La más seria de éstas es la endocarditis, la miocarditis y la pericarditis son menos comunes. La administración profiláctica de penicilina es necesaria para prevenir las manifestaciones de fiebre reumática. Después de la corea aguda, hasta la tercera parte de los pacientes persisten con riesgo de desarrollar valvulopatía cardiaca, principalmente estenosis mitral. Para detectar esta complicación potencial en forma temprana se recomiendan los ecocardiogramas seriados.
Tratamiento
El tratamiento es estrictamente sintomático, excepto por la profilaxis con penicilina de por vida. Al inicio se recomienda reposo en cama y se prescriben benzodiacepinas o antidepresivos para controlar la ansiedad, labilidad emocional y corea. Recientemente se han usado con éxito anticonvulsivantes GABAmiméticos (GABA significa -acido gama aminobutírico), como el valproato y el gabapentin, para tratar la corea.
ENFERMEDAD DE HUNTINGTON
Etiología
La enfermedad de Huntington es un padecimiento autosómico dominante uniformemente fatal que se caracteriza por disfunción motora, emocional y cognitiva progresivas, con edad de inicio entre los 35 y los 45 años (rango de tres a 70 años).62 Es causada por el gen huntingtin, una repetición poliglutamina inestable y que se expande (CAGn) en el brazo corto del cromosoma 4.63,64 La enfermedad de Huntington existe en todo el mundo y tiene una prevalencia de 10 casos por 100,000 habitantes.
Patogenia
La neuropatología de la EH consiste en atrofia cerebral diseminada, con afección más importante en el cuerpo estriado y la corteza cerebral. Al inicio la pérdida neuronal y la gliosis son máximas en el núcleo caudado y la corteza y otras estructuras subcorticales están menos afectadas.65-67 El mecanismo de muerte celular en la EH no es claro.68 Evidencias recientes indican que la producción excesiva de poliglutamina, dirigida por el gen huntingtin anormal, es un aspecto crucial en la patogenia de la enfermedad.68-70 Desde el punto de vista experimental, ratones transgénicos que portan la expansión CAGn forman masas proteináceas, intranucleares e insolubles dentro de las neuronas de los ganglios basales y la corteza.69,70 Un fenómeno similar puede explicar las ataxias espinocerebelosas,70 un grupo de padecimientos con CAGn en otros genes.
Manifestaciones clínicas
El diagnóstico de EH puede hacerse solo por los datos clínicos en pacientes que presentan corea y tienen una historia familiar positiva. Los síntomas de demencia y emocionales (v.gr., depresión, irritabilidad) pueden precedir y típicamente ocultar los síntomas motores porque son más incapacitantes. La evolución clínica puede ser hasta de 20 años.
En las fases tempranas de la enfermedad el examen físico suele revelar movimientos coreicos (i.e., aumento en el parpadeo o gesticulación) que progresan y llegan a afectar a múltiples partes del cuerpo. La corea típicamente alcanza su máximo en 10 años y es sustituida en forma gradual por inquietud muscular, hipertonía, distonía y temblor o mioclonías.71,72 En el seis a 10 porciento de los casos la EH puede manifestar características parkinsónicas más que corea (variante de Westphal). Esta variante suele presentarse a edad más temprana (i.e., inicia en personas de menos de 20 años de edad) y muestra con mayor frecuencia un patrón de herencia paterna.73
Otras alteraciones de movimiento en la EH incluyen bradicinesia y sacadas oculares lentas. Se altera la coordinación motora gruesa y la marcha, aumenta la torpeza y los pacientes se mueven con una marcha irregular, lenta y de base ancha. La coordinación motora fina e incluso el habla se alteran en forma severa. Los trastornos en la deglución pueden ocasionar neumonía por aspiración. Los pacientes progresan hasta un estado aquinético y prácticamente mudo.
Pruebas de laboratorio
El diagnóstico de EH puede confirmarse en la actualidad con pruebas genéticas.74 No es necesario realizar estas pruebas en las personas en las que existe confirmación genética o patológica de la enfermedad en otros familiares. Las medidas diagnósticas auxiliares incluyen IRM para determinar si existe atrofia selectiva del núcleo caudado, un dato típico que puede ser difícil de detectar.
Tratamiento
El tratamiento debe incluir un equipo multidisciplinario que pueda proporcionar apoyo social, médico, neuropsiquiátrico y genético a los pacientes y familiares durante el curso de la enfermedad. Se recomienda una revisión muy completa sobre los aspectos relacionados con consejo genético y pruebas presintomáticas.74 El tratamiento sintomático puede aliviar la corea, aunque este aspecto puede no ser el más incapacitante de la enfermedad. Los bloqueadores de la dopamina tienen eficacia moderada y pueden agravar las alteraciones de movimiento no coreicas de la EH, como la bradicinesia y la distonía. Los agentes antipsicóticos atípicos como la clozapina, el risperidone y la olanzapina son mejor tolerados y pueden también ayudar a controlar la corea. Las indicaciones para tratar la corea incluyen su interferencia con las actividades de la vida diaria y sociales. El tratamiento de la depresión es semejante al de la EP.
DISTONIA
La distonía es un término empleado para referirse tanto a un síntoma como a diversos trastornos hiperquinéticos. La distonía es uno de los trastornos de movimiento más comunes y también uno de ls diagnosticados y tratados en forma equívoca con más frecuencia. La prevalencia mundial es incierta porque los casos no se notifican, pero quizá exceda 300 millones, semejante a la de la esclerosis múltiple.75
Etiología
Las distonías pueden clasificarse de acuerdo con la edad de inicio, la región afectada del organismo y la etiología. Usando un esquema etiológico semejante al usado para la EP, las distonías pueden dividirse en síndromes primarios, secundarios, con distonía asociada, y padecimientos heredodegenerativos.
Las distonías primarias incluyen síndromes en los que la distonía es la manifestación clínica principal de la enfermedad. Aunque no se ha demostrado desde el punto de vista patológico, la afección parece relacionarse con los ganglios basales. La distonía primaria es uno de los pocos trastornos de movimiento con origen en los ganglios basales que no se asocian con trastornos cognitivos o psiquiátricos primarios. Sin embargo, esto no minimiza el impacto psicológico y social de la distonía, que puede ser muy importante por la verg[Yuml]enza, ansiedad y depresión asociadas.
El principal trastorno dentro de las distonías primarias es la distonía tipo 1, un padecimiento autosómico dominante hereditario que afecta principalmente a las familias de judíos Ashkenazi (hasta en el 90 porciento de los casos). La distonía tipo 1 puede ser generalizada o focal.75 La enfermedad tiene una penetrancia de alrededor del 30 porciento y es causada por un gen localizado en el cromosoma 9q34.
La distonía que responde a dopamina (DRD) se hereda también en forma autosómico dominante, con más frecuencia en mujeres. Se caracteriza por múltiples mutaciones en el gen para la GTP ciclohidrolasa I (GTP-CH), la enzima que limita la velocidad en la síntesis del cofactor de tirosina hidroxilasa tetrahidrobiopterina.76 La tirosina hidroxilasa es la enzima que limita la velocidad de síntesis de la dopamina. La DRD típicamente inicia en la niñez, comenzando en las piernas y diseminándose a los brazos. Son comunes las fluctuaciones diurnas. Algunos pacientes inician en épocas más tardías con síntomas parkinsónicos en lugar de distonía. Las características de la DRD son una respuesta excelente a dosis bajas de levodopa y un curso de la enfermedad no progresivo. La DRD puede ser confundida con parálisis cerebral atetoide, paraplegia espástica o parkinsonismo. Aunque rara, la DRD y sus otras formas fenotípicas responden tan bien a levodopa que se justifica una prueba terapéutica en todos los casos probables. La DRD parece ser un trastorno bioquímico puro sin cambios degenerativos en el cerebro.
Las distonías secundarias, que son causadas principalmente por medicamentos o factores ambientales, incluyen la distonía inducida por levodopa, la distonía aguda y tardía asociada con bloqueadores del receptor de dopamina, la distonía asociada con parálisis cerebral (PC atetoide), el traumatismo cerebral, la hipoxia cerebral y la lesión neurológica periférica. La distonía puede asociarse también con algunos estados infecciosos y posinfecciosos y con exposición tóxica a manganeso, cianuro y ácido 3-nitropropiónico.
Los síndromas con distonía asociada incluyen algunos trastornos metabólicos de aminoácidos, lípidos y mitocondriales encefalopáticos (v.gr., encefalomielopatía necrosante subaguda [enfermedad de Leigh]).
Los padecimientos heredodegenerativos con manifestaciones distónicas secundarias se presentan típicamente con características parkinsónicas predominantes. Estos incluyen enfermedad de Wilson y de Huntington, degeneración de los ganglios basales corticales, parálisis supranuclear progresiva, la forma Lubag de distonía-parkinsonismo (distonía tipo 3) y la EP.
Manifestaciones clínicas
Los datos distónicos incluyen contracciones musculares involuntarias que pueden ocurrir durante movimientos voluntarios (distonía de acción) o en reposo, y que causan la torsión de alguna parte del cuerpo y posturas anormales. La distonía se acompaña en ocasiones de un temblor tosco e irregular (temblor distónico) o incluso de un temblor postural típico. La contracción de los músculos antagonistas es una característica fundamental de la distonía, que la distingue de la corea, los tics y otras discinesias. La distonía se asocia también con el llamado flujo exagerado, la diseminación anomal de la activación a músculos diferentes de los requeridos para el movimiento que se intenta.
Las formas más comunes de distonía son las distonías focales, que pueden afectar (1) los párpados (blefaroespasmo), causando que se cierren en forma involuntaria, (2) el cuello y los hombros (distonía cervical), causando que el cuello se tuerza a un lado (tortícolis), hacia adelante (anterocolis) o hacia atrás (retrocolis), (3) la porción inferior de la cara y la mandíbula (distonía oromandibular), y (4) la laringe (distonía espasmódica), que ocasiona una voz tirante y de calidad irregular como resultado del cierre involuntario de las cuerdas vocales. La distonía focal puede afectar las manos y antebrazos durante el desempeño de actividades específicas, como escribir (calambre del escritor), teclear o tocar un instrumento musical (calambre del músico), o durante practicamente cualquier otra actividad que incluya acciones repetitivas de las manos (distonía ocupacional). Las distonías focales suelen confundirse con problemas psiquiátricos u ortopedicos. Comparando con las generalizadas, que comienzan en la infancia, las distonías focales tienden a presentarse en etapas más avanzadas de la vida. En los niños el primer signo de distonía generalizada se presenta como una distonía activa del pie. Después la distonía ocurre en reposo y eventualmente causa alteraciones posturales al diseminarse en forma ipsilateral y después a la extremidad contralateral.
Tratamiento
El tratamiento de la distonía es estrictamente sintomático y consiste en manejo médico y quirúrgico. Los detalles del mismo van más allá de los objetivos de este capítulo. En general, para tratar la distonía generalizada se usan dosis altas de anticolinérgicos, en ocasiones en combinación con neurolépticos o depletores de dopamina. Recientemente se ha empleado con éxito la cirugía estereotáctica (palidotomía y talamotomía) para tratar casos medicamente refractarios. Las distonías focales suelen responder poco a los medicamentos, pero mucho a las inyecciones de toxina botulínica en el grupo muscular afectado. La toxina botulínica actúa bloqueando la liberación de acetilcolina en la unión neuromuscular, causando debilidad dependiente de la dosis, lo que afecta la contracción espasmódica sin producir parálisis. Se requieren inyecciones repetidas cada dos a cinco meses.
ENFERMEDAD DE WILSON
Etiología
La enfermedad de Wilson (EW), o degeneración hepatolenticular, es un padecimiento autosómico recesivo del metabolismo del cobre con una prevalencia de uno por 30,000 habitantes.77 Aunque relativamente rara, es clínicamente importante porque es tratable y simula otros trastornos del movimiento. El gen de la enfermedad de Wilson, localizado en el cromosoma 13, codifica una proteína de 7.5 kb que se expresa en gran cantidad en el hígado y el cerebro.78 La proteína pertenece a una clase de ATPasas transportadoras de cobre dependientes de ATP. Múltiples mutaciones del gen de la EW dan origen al mismo fenotipo. La mayoría de las víctimas son heterocigotos compuestos que tienen diferentes mutaciones en cada alelo. Esto complica el diagnóstico molecular de la EW y limita el escrutinio a los familiares de pacientes conocidos.79,80
Patogenia
La enfermedad de Wilson es un trastorno de la ATPasa transportadora de cobre que causa menor incorporación hepática del cobre a la ceruloplasmina, menor excreción biliar de cobre, pérdida de los sitios de almacenamiento hepático, salida de cobre hacia el plasma y depósito patológico del mismo en el cerebro (principalmente en los ganglios basales), la córnea (produciendo el anillo de Kayser-Fleisher), el riñón (causando disfunción tubular), la médula ósea (con anemia y trombocitopenia) y el sistema musculoesquelético (ocasionando artritis y osteoporosis).80 Las crisis agudas (encefalopatía aguda con crisis convulsivas, crisis hemolítica y daño tubular agudo) son causadas por salida súbita de grandes cantidades de cobre hacia el torrente sanguíneo por iatrogenia (ver adelante) o después de infartos hepáticos o hepatitis aguda.
Manifestaciones clínicas
La edad de presentación varía entre tres y 60 años, con un pico de inicio alrededor de los 16 años. En un estudio de una serie extensa, el 54 porciento de los pacientes presentó manifestaciones neurológicas, 31 porciento disfunción hepática y hasta el 33 porciento manifestaciones psiquiátricas.77 La mayoría de los pacientes con alteraciones hepáticas son mujeres entre los ocho y los 16 años de edad, los hombres presentan con más frecuencia alteraciones neurológicas. El inicio puede variar desde falla hepática aguda fulminante hasta cirrosis insidiosa y leve. La EW puede ser la causa más común de disfunción hepática crónica en niños; todos los niños con esta manifestación deben ser estudiados buscando EW. Las principales manifestaciones hematológicas de la enfermedad incluyen anemia hemolítica, trombocitopenia (50 porciento de todos los pacientes con manifestaciones hematológicas) y neutropenia (30 porciento). Otras manifestaciones sistémicas son alteraciones del páncreas exócrino, hipoparatiroidismo, poliartritis, cálculos renales y cardiomiopatía por acúmulo miocárdico de cobre.
Las manifestaciones neurológicas de la EW son muy diversas, incluyendo varios trastornos del movimiento como distonías (65 porciento de los pacientes), rigidez o parkinsonismo (52 porciento), alteraciones de la postura y la marcha (42 porciento) y temblor (32 porciento). Sin embargo, la manifestación neurológica más común es la disartria (97 porciento), en ocasiones asociada con expresiones faciales alteradas (la clásica risa sardónica). Las manifestaciones psiquiátricas pleomórficas y de comportamiento de la EW incluyen irritabilidad, menor desempeño laboral, depresión y cambios en el estado de ánimo. Los trastornos afectivos pueden simular enfermedad bipolar y ocurren también síntomas psicóticos. Los problemas cognitivos pueden iniciar con pobre desempeño escolar y culminar en demencia.
Debido a la extrema variabilidad de las manifestaciones neuropsiquiáticas en la EW, es importante pensar en este diagnóstico en prácticamente todos los trastornos del movimiento que se presentan sin una etiología demostrable, en especial en jóvenes con sintomatología psiquiátrica.
Alteraciones de laboratorio
El diagnóstico puede hacerse en alrededor del 80 porciento de los casos al demostrar reducción en la ceruloplasmina sérica (EW, 0 a 200 mg/L, normal de 200 a 400 mg/L).77,81 Los pacientes con niveles de seruloplasmina normal se consideran heterocigotos. También está disminuido el cobre sérico (unido más libre), (EW 3 a 100 µmol/l, normal 11 a 24 µmol/L). La excreción aumentada de cobre en orina de 24 horas puede ser una medición más sensible que los niveles en suero (EW 100 a 1,000 µg en 24 horas, normal ² 40 µg en 24 horas), pero con este resultado, debe distinguirse a la EW de otros trastornos que producen colestasis. En los casos en los que los datos anteriores no son concluyentes, la prueba definitiva es la demostración de mayor contenido de cobre en la biopsia hepática (EW 200 a 3,000 µg/g de tejido seco, normal ² 50 µg/g de tejido seco). Casi todos los pacientes que tienen afección neurológica tienen los datos oftalmológicos clásicos del anillo de Kayser-Fleischer, o cataratas en girasol. Estos anillos representan depósitos de cobre en la membrana basal de la córnea [ver figura 6], y su detección se realiza con lámpara de hendidura. Los estudios de neuroimagen revelan pérdida del volumen cerebral, con dilatación ventricular en más del 95 porciento de las personas con afección neurológica, así como algunas neuropsiquiátricamente intactas. La IRM revela una señal hiperintensa única en las imágenes en T2 del núcleo lenticular, así como en las imágenes del tálamo, el puente mesocerebral y la circunvolución dentada en pacientes con enfermedad más avanzada [ver figura 5d]. Las imágenes en T1 pueden revelar lesiones de la sustancia blanca.82
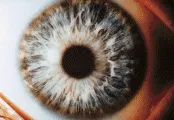
|
| Figura 6 |
| Anillos de Kayser-Fleischer |
Tratamiento
El tratamiento debe ser cuidadoso para evitar la encefalopatía aguda o la toxicidad hepática inducida por una movilización rápida del cobre.83,84 Los autores realizan una IRM antes de iniciar el tratamiento, incluso en pacientes asintomáticos. Este consiste en disminuir el cobre de la dieta y disminuir su absorción con acetato o sulfato de zinc (150 mg/día) o sulfuro de potasio (20 mg tid). Para movilizar los depósitos de cobre en los tejidos es necesario usar quelación del mismo con penicilamina. En los pacientes que no toleran la penicilamina (1 a 3 g/día por vía oral) o en los que empeoran al iniciar el tratamiento (10 a 40 porciento), otras alternativas incluyen el uso de trientine (con o sin dosis bajas de penicilamina) o dimercaprol y cloruro de cobalto (que rara vez se usa en la actualidad). Otras estrategias para disminuir el daño a los tejidos por el depósito de cobre son el uso de depuradores de radicales libres, como el mercaptodextrán.
El tratamiento quelante requiere vigilancia estrecha para lograr una excreción inicial de cobre en orina de 24 horas de 1.2 a 2.0 g/día. El tratamiento con penicilamina es mal tolerado en hasta el 30 porciento de los pacientes por efectos adversos alérgicos idiosincráticos, incluyendo condiciones autoinmunes. Contrario a la penicilamina, el tratamiento con zinc es bien tolerado. Puede aumentar la amilasa, lipasa y fosfatasa alcalina, y existen reportes aislados de efectos inmunosupresores. Cuando existe evidencia de progresión de la enfermedad a pesar del tratamiento dietético y farmacológico óptimos o cuando ocurre falla hepatica, el trasplante autólogo de hígado es el tratamiento más definitivo.85
Los síntomas neurológicos suelen responder mal al tratamiento. Pueden administrarse anticolinérgicos para tratar la distonía y el temblor, y administrarse dopaminomiméticos para la rigidez. Los síntomas psiquiátricos (ansiedad y depresión) se tratan en la forma habitual. La mejoría en los síntomas neuropsiquiátricos y otros puede retrasarse por meses o hasta por dos años después de iniciado el tratamiento quelante.
SINDROME DE TOURETTE
Etiología
El síndrome de Tourette (ST) es un padecimiento autosómico dominante en el que no se ha identificado el gen responsable. Tiene una expresión fenotípica muy amplia, penetrancia variable y expresión de género. Los criterios diagnósticos incluyen la presencia de múltiples tics motores y vocales que duran más de un año y aparecen antes de los 21 años. Los síntomas pueden ser suficientemente severos para justificar un diagnóstico médico y la alteración puede no asociarse con otros trastornos neurológicos o inducidos por medicamentos. Es importante la historia familiar, pero puede ser enmascarada por el fenotipo, que incluye sintomatología obsesivo-compulsiva (SOC) y trastornos de atención deficiente. La relación hombre-mujer para los tics es de 4:1, y la de la SOC es de 1:4. También pueden existir trastornos del aprendizaje y varios síndromes de comportamiento mal definidos.
Patogenia
La patogenia del síndrome de Tourette se desconoce, pero la evidencia farmacológica clínica y estudios limitados de autopsia sugieren que puede ser mediada por innervación hiperdopaminérgica del cuerpo estriado. Estudios morfométricos recientes que usan IRM del cerebro y estudios metabólicos usando tomografía de emisión de positrones sugieren la presencia de alteraciones anatómicas y funcionales en los circuitos corticoestriadopalidotalámico que se proyectan a la corteza prefrontal y a la circunvolución del cíngulo. La interrupción quirúrgica de estos circuitos (v.gr., tractotomía anterior y cingulotomías) puede ser útil para tratar la SOC y, en menor grado, los tics en casos seleccionados.
Manifestaciones clínicas
Un tic es una acción breve, estereotipada, rápida, repetitiva y aparentemente sin propósito que incluye uno o varios grupos musculares. Los tics tanto motores como vocales pueden ser simples (v.gr., parpadeo frecuente, retorcerse la nariz, toser, gruñir) o complejos (automutilación y coprolalia). Los tics pueden asociarse con experiencias sensoriales focales (tics sensoriales), que pueden preceder, acompañar o seguir a los movimientos o vocalización. En general los tics son la parte menos incapacitante del ST y pueden no requerir tratamiento. Durante los años escolares los trastornos en la atención, comportamiento (impulsividad e irritabilidad) y aprendizaje tienden a ser más incapacitantes. La SOC comienza al inicio de la adolescencia y puede ser muy incapacitante. Responde a clomipramina y a inhibidores selectivos de la recaptura de serotonina con menos frecuencia que el trastorno obsesivo-compulsivo sin ST. Los trastornos del sueño son frecuentes, pero mal comprendidos.
Tratamiento
El tratamiento del ST es difícil y debe realizarse por especialistas. Cuando los síntomas son complejos (v.gr., tics, SOC y deficiencia en la atención) los pediatras y neurólogos deben actuar junto con psiquiatras que guien y traten los síntomas de comportamiento. Los tics suelen responder a bloqueadores de dopamina (v.gr., prolixina, haloperidol y pimozide). Algunos niños son especialmente intolerantes a los efectos adversos de estos medicamentos, que varían de distonía aguda e inquietud motora (acatisia) a sedación diurna y fobias escolares. Reportes recientes sugieren que el neuroléptico atípico risperidone puede ser eficaz. Otros tratamientos para los tics incluyen clonidina (0.1 a 0.4 mg/día, como una cápsula o parche transdérmico) y clonacepam (0.5 a 4.0 mg/día).
Temblor fisiológico y esencial
El término temblor se usa para describir varios movimientos no relacionados que comparten una característica en común: oscilación de una extremidad o parte del organismo. Son ejemplos el templor en reposo del Parkinson y el temblor cerebeloso.
INCREMENTO DEL TEMBLOR FISIOLOGICO
El temblor fisiológico típicamente no es aparente, excepto cuando es exagerado por estrés, ansiedad o estimulantes. El miedo y otras formas de ansiedad situacional son ejemplos bien reconocidos y tratables de temblor fisiológico exagerado. Otras causas posibles de incremento del temblor incluyen agentes adrenérgicos, como la liberación endógena de epinefrina (por feocromocitoma, miedo o hipoglucemia) y liberación exógena de epinefrina, isoproterenol, terbutalina y teofilina. Los medicamentos que pueden inducir temblor fisiológico incluyen al litio, antidepresivos tricíclicos, estimulantes, valproato, esteroides, hormonas tiroideas y cafeína. La supresión de ciertos fármacos, como el alcohol, los opiáceos y las benzodiacepinas, así como el ejercicio excesivo (creando fatiga muscular) pueden causar mayor temblor fisiológico.
Tratamiento
El tratamiento incluye la eliminación o reducción de cualquiera de los agentes desencadenantes del temblor por medio de bloqueadores beta adrenérgicos (v.gr., propranolol), controlando la ansiedad y, cuando está indicado, tratando la endocrinopatía asociada. La ansiedad situacional puede controlarse administrando propranolol (20 a 60 mg) una hora antes del evento o situación desencadenante.
TEMBLOR ESENCIAL
El temblor esencial (TE) tiene una frecuencia de 6 a 8 Hz, es de origen central y con frecuencia tiene un patrón de herencia autosómico dominante.87 A diferencia de los pacientes con EP, los enfermos con TE pueden no tener otras características parkinsónicas y presentar solo movimiento aislado de la cabeza y temblor en la voz. Este último da al lenguaje una calidad vacilante. Los síntomas del TE se exacerban por los mismos factores que aumenta el temblor fisiológico.
Tratamiento
Característicamente, el temblor esencial responde rápido aunque en forma breve al alcohol (sin embargo, al disminuir la concentración sérica de alcohol el efecto de rebote puede empeorar el temblor). También responde a bloqueadores adrenérgicos beta (v.gr., propranolol) que suelen ser eficaces a largo plazo. Otros betabloqueadores útiles incluyen metoprolol, timolol, atenolol y sotalol. La primidona puede ser eficaz en dosis de 50 a 250 mg/día, que son menores que las requeridas para el tratamiento anticonvulsivante. Otros medicamentos que pueden ser benéficos incluyen benzodiacepinas (en especial clonacepam), fenobarbital y clonidina. La neurocirugía estereotáctica (talamotomía ventrolateral o estimulación cerebral profunda) es útil para los temblores severos refractarios a medicamentos.
Mioclonías
Características clínicas
Las mioclonías son movimientos súbitos, breves, involuntarios, como descargas. Pueden considerarse fragmentos de epilepsia, y tienen características electroencefalográficas que son semejantes a las descargas epilépticas focales. Las mioclonías positivas consisten en contracción muscular activa y las negativas en un lapso súbito dentro de la contracción que es clínicamente indistinguible de la asterixis. Las pruebas electrofisiológicas especializadas pueden localizar las descargas mioclónicas en la corteza somatosensorial, el tallo cerebral (el sistema activador reticular ascendente) y, en casos raros, la médula espinal. Como las crisis convulsivas, las descargas mioclónicas representan un signo específico de irritación del SNC con un diagnóstico diferencial amplio [ver tabla 4].86
Las descargas mioclónicas pueden ser focales, segmentarias o generalizadas. Cuando son focales sugieren una patología focal, que varía de epilepsia a encefalitis focal [ver tabla 4]. Pueden ser espontáneas, sensibles a estímulos (reflejas) o relacionarse a actividades (de acción). Cuando son generalizadas pueden asociarse con epilepsia o con una encefalopatía estática y progresiva. Para disminuir las posibilidades diagnósticas durante el examen es importante buscar signos de encefalopatía, demencia, patología retininana, pérdida de la audición, miopatía, neuropatía y ataxia.
Diagnóstico diferencial
El diagnóstico diferencial de las mioclonías consiste en determinar si las contracciones son causadas por una enfermedad médica, exposición a un fármaco o toxina, o un trastorno primario del sistema nervioso central.89 Un elemento importante en el diagnóstico diferencial es si las mioclonías son epilépticas o no.90,91 La edad de inicio es muy importante porque las formas progresivas más comunes de epilepsia que son causadas por procesos degenerativos tienden a ocurrir en el grupo de edad pediátrico.
Pruebas de laboratorio
El escrutinio de laboratorio básico debe incluir una biometría hemática, química sanguínea y estudio toxicológico. Están indicados los estudios de imagen cerebral y la electroencefalografía cuando la causa de la mioclonía no es inmediatamente aparente. Cuando la clínica lo justifica deben realizarse estudios metabólicos especializados para descartar trastornos inflamatorios sistémicos, endocrinopatías, trastornos del metabolismo del cobre (v.gr., EW y enfermedad de Menke), deficiencias de enzimas seleccionadas y trastornos de almacenamiento de lisosomas [ver tabla 4 ]. Un subgrupo de estos padecimientos, como algunos de los trastornos de la fosforilación oxidativa mitocondrial (ver adelante) y algunas de las ataxias espinocerebelosas pueden diagnosticarse en la actualidad con pruebas genéticas.
Tratamiento
El tratamiento de las mioclonías es estrictamente sintomático. Los medicamentos de uso común incluyen clonacepam (2 a 12 mg/día), valproato de sodio (600 a 3,000 mg/día), primidona (500 a 1,500 mg/día) y, en Europa, piracetam (6 a 20 mg/día). Los inhibidores selectivos de la recaptura de serotonina y el 5-hidroxitriptofano (con carbidopa) pueden ayudar en algunas formas específicas de mioclonía (la mioclonía reticular refleja) pero puede agravar otras. Otra clase especial es la mioclonía del paladar, que responde a trihexifenidilo (6 mg/día). El tratamiento de los síndromes epilépticos asociados con trastornos mioclónicos no será analizado en este capítulo.
MIOCLONIAS EN LA PRACTICA MEDICA GENERAL
En la práctica médica general la forma más común de mioclonía es resultado de trastornos metabólicos, en los que las mioclonías negativas o asterixis tienden a dominar el cuadro clínico [ver tabla 4]. La etiología de las mioclonías incluye también encefalitis viral, que típicamente se presenta como mioclonías positivas y negativas. Son ejemplos las encefalitis asociadas con virus herpes simple y arbovirus, así como las asociadas con síndromes posinfecciosos (v.gr., panencefalitis esclerosante subaguda y encefalitis letárgica).
Otras formas comunes de mioclonías observadas en la práctica médica general incluyen mioclonías inducidas por medicamentos y mioclonías secundarias a suspensión de fármacos. Los medicamentos que inducen mioclonías incluyen carbonato de litio, antidepresivos tricíclicos e inhibidores selectivos de la recaptura de serotonina (ISRS). La coadministración de ISRS e inhibidores de la MAO A o MAO B pueden causar un síndrome hiperserotoninérgico que típicamente inicia con mioclonías y cambios en el estado mental. Los agentes antinfecciosos asociados en ocasiones con mioclonías incluyen a las penicilinas y las cefalosporinas [ver tabla 3]. En la anestesia, los hipnóticos no barbitúricos de acción corta (v.gr., etomidato) pueden causar también mioclonías. Los pacientes con EP que están tomando levodopa en ocasiones desarrollan mioclonías.
La sobredosis o intoxicación con anticonvulsivantes, antihistamínimos o hipnosedantes pueden causar mioclonías, con o sin crisis generalizadas. Otros medicamentos incluyen los antineoplásicos como el clorambucil, los antinflamatorios no esteroides (AINE) cuando se administran a pacientes con insuficiencia renal y el bismuto. La exposición extensa a las siguientes toxinas también se asocia con mioclonías: metilbromuro, mercurio orgánico, tetraetilo de plomo (gasolina), estricnina y DDT. La supresión de alcohol, benzodiacepinas u opiáceos causa mioclonías con frecuencia. Por último, las mioclonías pueden ser una secuela de la lesión cerebral global por traumatismos, anoxia-isquemia, golpe de calor y electrocución.92
Los trastornos demenciales asociados con mioclonías incluyen padecimientos asociados a priones, como la enfermedad de Creutzfeld-Jakob. Además, un pequeño subgrupo de pacientes con enfermedad de Alzheimer (10 a 15 porciento) y un gran número de pacientes con enfermedad difusa de cuerpos de Lewy presentan mioclonías. Estos padecimientos pueden evolucionar de mioclonías focales a formas más generalizadas.93
Algunos otros trastornos representan problemas genéticos primarios poco comunes con sintomatología compleja asociada [ver tabla 4].94,95 Además de controlar las mioclonías y crisis consulsivas, el tratamiento para estas condiciones es muy limitado.
Trastornos del movimiento inducido por medicamentos
Los trastornos del movimiento inducidos por medicamentos son un grupo importante de condiciones iatrogénicas y casi siempre reversibles que se presentan con frecuencia en la práctica clínica general. La frecuencia de los trastornos del movimiento inducidos por medicamentos dependen de la naturaleza del compuesto, la edad de la persona afectada y otros factores de riesgo (ver adelante).96
La mayoría de los trastornos del movimiento relacionados con medicamentos se asocian con agentes que afectan en forma directa o indirecta la trasmisión dopaminérgica central. Las categorías de fármacos incluyen los estimulantes del SNC, el precursor de dopamina levodopa, los agonistas dopaminérgicos de acción directa y los bloqueadores del receptor central de dopamina. Los tipos de movimientos producidos por estos compuestos pueden ser agudos, subagudos o tardíos, como en el caso de la discinesia tardía o distonía tardía. Los mecanismos de los trastornos de movimiento agudos y subagudos parecen ser extensiones idiosincráticas de la acción del compuesto. Por el contrario, el mecanismo de la discinesia tardía no se ha aclarado.
Alrededor de la tercera parte de los pacientes con discinesia tardía remiten a los tres meses de suspender el tratamiento neuroléptico. En la mayoría de los pacientes los movimientos remiten en forma gradual en cinco años. Los pacientes con riesgo de discinesia tardía permanente incluyen los ancianos, los desdentados y los que sufren disfunción orgánica cerebral. Los pacientes con enfermedades afectivas tienen más riesgo de desarrollar discinesia tardía que los esquizofrénicos.
TRASTORNOS DEL MOVIMIENTO AGUDOS INDUCIDOS POR FARMACOS
Las reacciones de esta categoría incluyen la distonía aguda en respuesta a bloqueadores centrales de dopamina. Parecen ser más frecuentes en varones jóvenes y en pacientes que reciben medicamentos como litio, bloqueadores de los canales del calcio e ISRS. La distonía aguda puede tratarse con la administración parenteral de anticolinérgicos (benzotropina o difenhidramina) o benzodiacepimas (loracepam o diacepam). Otros trastornos de movimiento agudos incluyen discinesias, comportamientos estereotipados en respuesta a neurolépticos y tics, que pueden ocurrir en respuesta a estimulantes del SNC como metilfenidato, dextroanfetamina y pemoline.
TRASTORNOS DEL MOVIMIENTO SUBAGUDOS INDUCIDOS POR FARMACOS
Es probable que la reacción más común de este tipo sea la acatisia inducida por neurolépticos.97 La acatisia es un estado de inquietud motora que consiste en una necesidad imperiosa de moverse. El movimiento tiende a aliviar los síntomas en forma temporal. La inquietud parece estar mediada por el bloqueo del receptor de dopamina mesolímbico. Desde el punto de vista sintomático, esto puede relacionarse con el síndrome de piernas inquietas. El tratamiento consiste en retirar el agente agresor. Cuando esto no es posible los síntomas pueden disminuir con benzodiacepinas, anticolinérgicos, betabloqueadores y, en algunos casos, agentes dopaminérgicos.
TRASTORNOS DE MOVIMIENTO TARDIOS
Manifestaciones clínicas
Los trastornos de movimiento tardíos son causados principalmente por exposición crónica (mayor de tres meses) a bloqueadores centrales de dopamina.98 Los movimientos son principalmente coreicos y se conocen como discinesia tardía (DT). Al inicio se afectan los músculos del cráneo (boca, labios y lengua en particular), y después el tronco y las extremidades. En los casos más avanzados una persona puede sufrir movimiento continuo de la cabeza, rotación de la pelvis y movimientos finos de los dedos de manos y pies. En raros casos la discinesia tardía puede afectar el diafragma y causar falla respiratoria. Los pacientes con distonía tardía presentan contracciones distónicas axiales principalmente, más que apendiculares. Otros síndromes tardíos incluyen la acatisia tardía, los tics tardíos e incluso temblor tardío.99
Tratamiento
El mejor tratamiento para la DT es la prevención. Se recomienda el uso juicioso de neurolépticos por el periodo más corto posible. Los neurolépticos atípicos se asocian con un riesgo significativamente menor de causar DT que los neurolpépticos convencionales, para los que el riesgo varía de 13 a 20 porciento. El menor riesgo asociado a los neurolépticos atípicos puede atribuirse a su mayor afinidad por los receptores de dopamina mesolímbicos D4 y D3, más que los receptores estriados D2. Los neurolépticos atípicos incluyen la clozapina, el resperidone y la olanzapina, recién autorizada.
Cuando no es posible suspender el agente que causa la DT, debe considerarse cambiar los neurolépticos tradicionales por atípicos.99 Si el paciente está tomando también estimulantes y anticolinérgicos, el eliminarlos puede aliviar la discinesia. La reserpina es el tratamiento de elección para la discinesia tardía coreiforme. La dosis inicial debe ser de 0.125 mg/día y puede aumentarse lentamente hasta 6 mg/día, según se requiera. Los ancianos tienen menos probabilidad de tolerar el tratamiento por hipotensión ortostática. El uso a largo plazo de reserpina se asocia también con un incremento de 15 porciento en la incidencia de depresión.
Otra estrategia de tratamiento consiste en estimular el sistema GABAérgico usando baclofen (40 a 80 mg/día), clonacepam (1 a 8 mg/día) o ácido valproico (1 a 3 g/día). Las estrategias GABAérgicas son especialmente útiles en pacientes con discinesia tardía que pueden beneficiarse del tratamiento anticolinérgico y de inyecciones de toxina botulínica en los músculos más afectados. La buspirona (40 a 60 mg/día) es un tratamiento GABAmimético que ha demostrado ser útil en algunos casos.
Este capítulo fue patrocinado en parte por una beca del Centro de Excelencia de la Emory University School of Medicine de la American Parkinson's Disease Association, de Nueva York.
Figuras 1 y 2 Seward Hung
Figura 3 Cortesía del Dr. Thomas M. Aaberg, Sr., Servicio de Oftalmología, Emory University School of Medicine.
Bibliografía
- Alexander G, Crutcher MD: Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 13:266, 1990 [PMID 1695401]
- Ashby P, Andrews C, Knowles L, et al: Pyramidal and extrapyramidal control of tonic mechanisms in the cat. Brain 95:21, 1972 [PMID 5023088]
- DeLong M: Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13:281, 1990
- Bergman H, Wichmann T, DeLong MR: Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249:1436, 1990 [PMID 2402638]
- Laitinen L, Bergenheim HT, Hariz MI: Leksell's posteroventral pallidotomy in the treatment of Parkinson's disease. J Neurosurg 76:53, 1992 [PMID 1727169]
- Grafton S, Waters C, Sutton J, et al: Pallidotomy increases activity of motor association cortex in Parkinson's disease: a positron emission tomography study. Ann Neurol 37:776, 1995
- Albin RL, Reiner A, Anderson KD, et al: Striatal and nigral neuron subpopulations in rigid Huntington's disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol 27:357, 1990 [PMID 1972318]
- Albin RL, Reiner A, Anderson KD, et al: Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol 31:425, 1992 [PMID 1375014]
- Alexander G, Crutcher MD, DeLong MR: Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, "prefrontal" and "limbic" functions. Prog Brain Res 85:119 1990
- Parkinson J: An Essay on the Shaking Palsy. Sherwood, Neely & Jones, London, 1817
- Martilla R: Epidemiology. Handbook of Parkinson's Disease. Koller W, Ed. Marcel Dekker, New York, 1987
- Johnson W, Hodge SE, Duvoisin RC: Twin studies and the genetics of Parkinson's disease: a reappraisal. Mov Disord 5:187, 1990 [PMID 2388635]
- Calne D, Shoenberg BS, Martin W, et al: Familial Parkinson's disease: possible role of environmental factors. Can J Neurol Sci 14:303, 1987
- Polymeropoulos MH, Lavedan C, Leroy E, et al: Mutation in the a -synuclein gene identified in families with Parkinson's disease. Science 276:2045, 1997 [PMID 9197268]
- Forno LS: The Lewy body in Parkinson's disease. Adv Neurol 45:35, 1986
- Olanow CW, Hauser RA, Gauger L, et al: The effect of deprenyl and levodopa on the progression of Parkinson's disease. Ann Neurol 38:771, 1995 [PMID 7486869]
- Ahlskog JE, Uitti RJ, Low PA, et al: Levodopa and deprenyl treatment effects on peripheral indices of oxidant stress in Parkinson's disease. Neurology 46:796, 1996 [PMID 8618686]
- Hughes A, Ben-Shlomo Y, Daniel SE, et al: What features improve the accuracy of clinical diagnosis in Parkinson's disease: a clinicopathologic study. Neurology 42:1142, 1992 [PMID 1603339]
- Rajput A: Prevalence of dementia in Parkinson's Disease. Parkinson's Disease: Neurobehavioral Aspects. Huber S, Cummings JL, Eds. Oxford University Press, New York, 1992, p 119
- Cummings J: Intellectual impairment in Parkinson's disease: clinical, pathologic, and biochemical correlates. Journal of Geriatric Psychiatry and Neurology 1:24, 1988
- Taylor A, Saint-Cyr JA, Lang AE, et al: Parkinson's disease and depression. Brain 109:279, 1986
- Starkstein S, Mayberg HS, Leiguarda R, et al: A prospective longitudinal study of depression, cognitive decline and physical impairments in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 55:377, 1992 [PMID 1602311]
- Menza M, Rosen RC: Sleep in Parkinson's disease: the role of depression and anxiety. Psychosomatics 36:262, 1995 [PMID 7638313]
- Comella C, Tanner CM, Ristanovic RK: Polysomnographic sleep measures in Parkinson's disease patients with treatment-induced hallucinations. Ann Neurol 34:710, 1993 [PMID 8239565]
- Bliwise D, Watts RL, Watts N, et al: Disruptive nocturnal behavior in Parkinson's disease and Alzheimer's disease. J Geriatr Psychiatry Neurol 8:107, 1995 [PMID 7794473]
- Cotzias G, Papvasiliou PS, Gellene R: Modification of parkinsonism: chronic treatment with L-dopa. N Engl J Med 280:337, 1969 [PMID 4178641]
- Caraceni T, Scigliano G, Musicco M: The occurrence of motor fluctuations in parkinsonian patients treated long term with levodopa: role of early treatment and disease progression. Neurology 41:380, 1994
- Blin J, Bonnet AM, Agid Y: Does levodopa aggravate Parkinson's disease? Neurology 38:1410, 1988
- Ahlskog JE: Treatment of early Parkinson's disease: are complicated strategies justified? Mayo Clinic Proc 71:659, 1996
- Jenner P, Schapira AHV, Marsden CD: New insights into the cause of Parkinson's disease. Neurology 42:2241, 1992 [PMID 1461374]
- Olanow C: A radical hypothesis for neurodegeneration. Trends Neurosci 16:439, 1993
- Pahwa R, Koller WC: Treatment of Parkinson's disease with controlled-release carbidopa/L-DOPA. Adv Neurol 69:487,1996 [PMID 8615169]
- Block G, Liss C, Reines S, et al: Comparison of immediate-release and controlled-release carbidopa/levodopa in Parkinson's disease. Eur Neurol 37:23, 1997 [PMID 9018028]
- Birkmayer W, Knoll J, Riederer P, et al: Increased life expectancy resulting from addition of L-deprenyl to Madopar treatment in Parkinson's disease: a longterm study. Journal of Neural Transmission 64:113, 1985 [PMID 3935752]
- Nutt J, Woodard WR, Beckner RM, et al: Effect of peripheral catechol-O-methyltransferase inhibition on the pharmacokinetics and pharmacodynamics of levodopa in parkinsonian patients. Neurology 44:913, 1994 [PMID 8190296]
- Weiner WJ, Factor SA, Sanchez-Ramos JR, et al: Early combination therapy (bromocriptine and levodopa) does not prevent motor fluctuations in Parkinson's disease. Neurology 43:21, 1993 [PMID 8423888]
- Greenamyre JT, O'Brien CF: N-methyl-D-aspartate antagonists in the treatment of Parkinson's disease. Arch Neurol 48:977, 1991 [PMID 1835370]
- Uitti RJ, Rajput AH, Ahlskog JE, et al: Amantadine treatment is an independent predictor of improved survival in parkinsonism. Neurology (in press)
- Effect of deprenyl on the progression of disability in early Parkinson's disease. Parkinsonism Study Group. N Engl J Med 321:1364, 1989
- Tetrud J, Langston JW: The effect of deprenyl (selegiline) on the natural history of Parkinson's disease. Science 245:519, 1989 [PMID 2502843]
- Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. Parkinsonism Study Group. N Engl J Med 328:176, 1993
- Brannon T, Yahr MD: Comparative study of selegiline plus L-dopa-carbidopa versus L-dopa-carbidopa alone in the treatment of Parkinson's disease. Ann Neurol 37:95, 1995
- Impact of deprenyl and tocopherol treatment on Parkinson's disease in DATATOP subjects not requiring levodopa. Parkinsonism Study Group. Ann Neurol 39:29, 1996
- Lees A: Comparison of therapeutic effects and mortality data of levodopa and levodopa combined with selegiline in patients with early, mild Parkinson's disease. BMJ 311:1602, 1995
- Marsden C, Parkes JD: On-off effects in patients with Parkinson's disease on chronic levodopa therapy. Lancet 1:292, 1981
- Kurlan R, Rothfield K, Woodward WR, et al: Erratic gastric emptying of levodopa may cause "random" fluctuations of parkinsonian mobility. Neurology 38:419, 1988 [PMID 3126408]
- Cummings J: Behavioral complications of drug treatment of Parkinson's disease. J Am Geriatr Soc 39:708, 1991
- Wolters EC, Jansen ENH, Tuynman-Qua HG, et al: Olanzapine in the treatment of dopaminomimetic psychosis in patients with Parkinson's disease. Neurology 47:1085, 1996
- Balldin J, Eden S, Granerus A-K, et al: Electroconvulsive therapy in Parkinson's syndrome with "on-off" phenomenon. Journal of Neural Transmission 47:11, 1980 [PMID 7359119]
- Lozano A, Lang AE, Galvez-Jiminez N, et al: Effects of GPi pallidotomy on motor function in Parkinson's disease. Lancet 346:1383, 1995 [PMID 7475819]
- Dogali M, Fazzinni E, Koloday E, et al.: Stereotactic ventral pallidotomy for Parkinson's disease. Neurology 45:753, 1995
- Baron MS, Vitek JL, Bakay RA, et al: Treatment of advanced Parkinson's disease by posterior GPi pallidotomy: 1-year results of a pilot study. Ann Neurol 40:355, 1996 [PMID 8797525]
- Hardie R, Lees AJ: Neuroleptic-induced Parkinson's syndrome: clinical features and results of treatment with levodopa. J Neurol Neurosurg Psychiatry 51:850, 1988 [PMID 2900293]
- Jimenez-Jimenez FX, Ort'-Pareja M, Ayusi-Peralta L, et al: Drug-induced parkinson isms in a movement disorders unit: a four-year survey. Parkinsonism and Related Disorders 2:145, 1996
- Tolosa E, Duvoisin R, Cruz-Sanchez FF: Progressive Supranuclear Palsy: Diagnosis, Pathology, and Therapy. Springer-Verlag, New York, 1994
- Quinn N: Multiple system atrophy: the nature of the beast. J Neurol Neurosurg Psychiatry June(suppl):78, 1989
- Lang A, Riley DE, Bergeron C: Cortico-basal ganglionic degeneration. Neurodegenerative Diseases. Calne D, Ed. WB Saunders Co, Philadelphia, 1994
-
Thompson P, Marsden CD: Gait disorder of subcortical
arteriosclerotic encephalopathy: Binswanger's disease. Move Disord 2:1,
1987
-
Ditter S, Mirra SS: Neuropathologic and clinical features
of Parkinson's disease in Alzheimer's disease patients. Neurology 37:754,
1987 [PMID
3033544]
-
Mega M, Masterman DL, Benson F, et al: Dementia with
Lewy bodies: reliability and validity of clinical and pathologic criteria.
Neurology 47:1403, 1996 [PMID
8960718]
-
Shoulson I: On chorea. Clin Neuropharmacol 9(suppl
2): S85, 1986
-
Folstein SE: Huntington's Disease: a Disorder of Families.
Johns Hopkins University Press, Baltimore, 1990
-
A novel gene containing a trinucleotide repeat that
is expanded and unstable on Huntington's disease chromosomes. Huntington's
Disease Collaborative Research Group. Cell 72:971, 1993
-
Skraastad MI, deRoou KE, deKoning-Gans PAM: Defining
the proximal border of the Huntington's disease candidate region by multipoint
recombination analyses. Genomics 16:599, 1993
-
Myers R, Vonsattel J: Clinical and neuropathologic
assessment of severity in Huntington's disease. Neurology 38:341, 1988
[PMID
2964565]
-
Braak H, Braak E: Allocortical involvement in Huntington's
disease. Neuropathol Appl Neurobiol 18:539, 1992
-
Storey E, Kowall NW, Finn SF, et al: The cortical lesion
of Huntington's disease: further neurochemical characterization and reproduction
of some of the histological and neurochemical features by N-methyl-D-aspartate
lesions of rat cortex. Ann Neurol 32:526, 1992 [PMID
1280937]
-
Young AB: Huntington's disease: lessons from and for
molecular neuroscience. Neuroscientist 1:1, 1994
-
Davies SW, Turmaine M, Cozens BA, et al: Formation
of neuronal intranuclear inclusions underlies the neurological dysfunction
in mice transgenic for the HD mutation. Cell 90:537, 1977
-
Scherzinger E, Lurz R, Turmaine M, et al: Huntingtin-encoded
polyglutamine expansions form amyloid-like aggregates in vitro and in vivo.
Cell 90:549, 1997 [PMID
9267034]
-
Shoulson I: Huntington's disease: functional capacities
in patients treated with neuroleptic and antidepressant drugs. Neurology
31:333, 1981
-
Martin JB, Gusella JF: Huntington's disease pathogenesis
and management. N Engl J Med 315:1267, 1986
-
Duyao M, Ambrose C, Myers R, et al: Trinucleotide repeat
length instability and age of onset in Huntington's disease. Nat Genet
4:387, 1993 [PMID
8401587]
-
Hersch S, Jones R, Koroshetz W, et al: The neurogenetics
genie: testing for the Huntington's disease mutation. Neurology 44:1369,
1994 [PMID
7914682]
-
Bressman S, DeLeon D, Kramer PL, et al: Dystonia in
Ashkenazi Jews: clinical characterization of a founder mutation. Ann Neurol
37:140, 1995
-
Furukama Y, Shimadzu M, Rajput AH, et al: GTP-cyclohydrolase
I gene mutations in heriditary progressive and dopa-responsive dystonia.
Ann Neurol 39:609, 1996
-
Brewer JB, Yuzbasiyan-Gurkan V: Wilson's disease. Medicine
(Baltimore) 71:139, 1992
-
Petrukhin K, Fischer SG, Pirastu M, et al: Mapping,
cloning and genetic characterization of the region containing the Wilson
disease gene. Nat Genet 5:338, 1993 [PMID
8298640]
-
Thomas G, Forbes JR, Roberts EA, et al: The Wilson
disease gene: spectrum of mutations and their consequences. Nat Genet 9:210,
1995 [PMID
7626145]
-
Petrukhin K, Lutsenko S, Chernov I, et al: Characterization
of the Wilson disease gene encoding a P-type copper transporting ATPase:
genomic organization, alternative splicing, and structure/function predictions.
Hum Mol Genet 3:1647, 1994 [PMID
7833924]
-
McCullough AJ, Fleming CR, Thistle JL, et al: Diagnosis
of Wilson's disease presenting as fulminant hepatic failure. Gastroenterology
84:161, 1983 [PMID
6847842]
-
Prayer L, Wumberger D, Kramer J, et al: Cranial MRI
in Wilson's disease. Neuroradiology 32:211, 1990 [PMID
2215906]
-
Scheinberg IH, Sternlieb I: Wilson's Disease. WB Saunders
Co, Philadelphia, 1984
-
Walshe JM, Yealland M: Chelational treatment of neurological
Wilson's disease. Quarterly Journal of Medicine 86:197, 1993 [PMID
8369040]
-
Bellary S, Hassanein T, Van Thiel DH: Liver transplantation
for Wilson's disease. Hepatology 23:373, 1995
-
Kurlan R: Handbook of Tourette's Syndrome and Related
Tic and Behavioral Disorders. Marcel Dekker, New York, 1993
-
Elble R, Koller WC: Tremor. Johns Hopkins University
Press, Baltimore, 1990
-
Fahn S, Marsden CD, Van Woert MH: Definition and classification
of myoclonus. Arch Neurol 43:1, 1986
-
Caviness JN: Myoclonus. Mayo Clin Proc 71:679, 1996
-
Aicardi J: Myoclonic epilepsies of infancy and childhood.
Adv Neurol 43:11, 1986
-
Berkovic SF, Andermann F, Carpenter S, et al: Progressive
myoclonus epilepsies: specific causes and diagnosis. N Engl J Med 315:296,
1986 [PMID
3088452]
-
Fahn S: Posthypoxic action myoclonus: review of the literature
and report of two new cases with response to valproate and estrogen. Adv
Neurol 26:49, 1979
-
Hallett M, Wilkins DE: Myoclonus in Alzheimer's disease
and minipolymyoclonus. Adv Neurol 43:399, 1986
-
So N, Berkovic S, Andermann F, et al: Myoclonus epilepsy
and ragged-red fibers (MERF): 2. Electrophysiological studies and comparison
with other progressive myoclonus epilepsies. Brain 112:1261, 1989
-
Marsden CD, Harding AE, Obeso JA, et al: Progressive
myoclonic ataxia (the Ramsay Hunt syndrome). Arch Neurol 47:1121, 1990
[PMID
2121121]
-
Gershanik OS: Drug-induced movement disorders. Current
Opinion in Neurology and Neurosurgery 6:369, 1993
-
Fleischacker WW, Roth SD, Kane JM: The pharmacologic
treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol 10:12,
1990
-
Jeste D, Wyatt, RJ: Changing epidemiology of tardive dyskinesia.:
an overview. Am J Psychiatry 138:297, 1981
- Kang UJ, Burke RE, Fahn S: Natural history and treatment of tardive dystonia. Mov Disord 1:193, 1986