Dermatología
⭳ Abrir artículo (PDF)1.2 MBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
I MANIFESTACIONES CUTANEAS DE LAS ENFERMEDADES SISTEMICAS
- Enfermedades cardiopulmonares y vasculares
- SARCOIDOSIS
- VASCULITIS GRANULOMATOSA
- HIPERLIPOPROTEINEMIA
- ENFERMEDAD DE KAWASAKI
- SEUDOXANTOMA ELASTICO
- FIEBRE REUMATICA
- SINDROME DE LAS UÑAS AMARILLAS
- Enfermedades endócrinas
- Enfermedades gastrointestinales
- SINDROME CARCINOIDE
- ENFERMEDAD INFLAMATORIA DEL INTESTINO
- ENFERMEDAD DE CROHN METASTASICA
- TRASTORNOS CUTANEOS CON COMPLICACIONES GASTROINTESTINALES
- Enfermedades hematológicas
- Inmunodeficiencias
- Enfermedades infecciosas
- ENDOCARDITIS INFECCIOSA
- SINDROME DE CHOQUE TOXICO
- SINDROME DE PIEL ESCALDADA POR ESTAFILOCOCO
- FASCITIS NECROSANTE
- MENINGOCOCCEMIA
- FIEBRE ESCARLATINA
- INFECCION POR VIBRIO
- ENFERMEDAD DE LYME
- FIEBRE MANCHADA DE LAS MONTAÑAS ROCOSAS
- Enfermedades neurológicas
- SINDROME DE NEVOS DE CELULAS BASALES
- SINDROME DE NEVO EPIDERMICO
- INCONTINENCIA DE PIGMENTO
- NEUROFIBROMATOSIS
- SINDROME DE SNEDDON
- ESCLEROSIS TUBEROSA
- Enfermedades renales
- Enfermedades reumatológicas
I MANIFESTACIONES CUTANEAS DE LAS ENFERMEDADES SISTEMICAS
DR. MARK LEBWOHL
Las manifestaciones cutáneas de las enfermedades sistémicas son tan numerosas y diversas que es imposible abarcarlas en un solo capítulo, incluso en forma somera. En lugar de ello, este capítulo revisa manifestaciones cutáneas claves de enfermedades sistémicas que deben ser detectadas por la mayoría de los médicos y destaca los adelantos recientes en el diagnóstico y tratamiento de estos padecimientos. Para mayor descripción de las enfermedades específicas, incluyendo sus manifestaciones cutáneas, los lectores deben consultar los capítulos dedicados a estos padecimientos.
En muchos de los trastornos presentados en este capítulo, la evaluación y tratamiento del padecimiento sistémico subyacente son indispensables para tener una evolución favorable. Por ejemplo, el dato de sarcoidosis cutánea obliga a buscar sarcoidosis sistémica. En otros padecimientos, por ejemplo en la epidermolisis bulosa distrófica recesiva, el tratamiento del trastorno cutáneo es clave para el manejo de la enfermedad sistémica.
Enfermedades cardiopulmonares y vasculares
SARCOIDOSIS
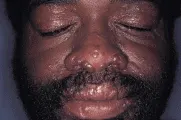
Las manifestaciones cutáneas de la sarcoidosis son tan variadas como sus manifestaciones sistémicas. Las pápulas alrededor de los ojos y la nariz son las más características. El término lupus pernio se refiere a granulomas no caseosos que provocan placas translúcidas y violáceas en los oídos, mejillas y nariz [ver figura 1]. Puede ocurrir afección del hueso subyacente y el diagnóstico se realiza por biopsia de piel. El tratamiento tradicional consiste en esteroides intralesionales y se han usado con éxito antipalúdicos y metotrexate. En algunos pacientes con sarcoidosis ocurre eritema nodoso, caracterizado por nódulos profundos, dolorosos y eritematosos en las extremidades inferiores. El lupus pernio se asocia con una evolución más crónica, mientras que el eritema nodoso indica una enfermedad aguda y más benigna.1
VASCULITIS GRANULOMATOSA
Granulomatosis de Wegener
La granulomatosis de Wegener se asocia con signos mucocutáneos tanto distintivos como no específicos. La púrpura palpable es uno de los datos cutáneos más comunes, aunque también se han descrito úlceras, pápulas, nódulos y bulas. Además de los síntomas de vías respiratorias superiores y pulmonares , la presencia de deformidad de la nariz en silla de montar, ulceraciones nasales y perforación septal debe sugerir el diagnóstico de granulomatosis de Wegener. El diagnóstico definitivo se realiza demostrando vasculitis con granulomas no caseosos en un paciente con afección de vías respiratorias superiores e inferiores y glomerulonefritis. Con frecuencia existen anticuerpos anticitoplasma de neutrófilo.
Granulomatosis linfomatoide
La granulomatosis linfomatoide puede asociarse con lesiones cutáneas inespecíficas, incluyendo máculas eritematosas, pápulas, placas, nódulos y úlceras. Este trastorno se distingue clínicamente de la granulomatosis de Wegener por la ausencia de afección de las vías respiratorias superiores. El diagnóstico se establece al demostrar un infiltrado necrosante granulomatoso con células linfoides atípicas alrededor de los vasos sanguíneos. Este trastorno se ha atribuido a proliferación clonal de las células B que induce una respuesta de células T.2
Síndrome de Churg-Strauss
El síndrome de Churg-Strauss, o angeítis granulomatosa alérgica, se presenta con más frecuencia como asma y eosinofilia; sin embargo, se desarrollan lesiones cutáneas en hasta el 40 por ciento de los pacientes. Los datos más comunes consisten en púrpura palpable simétrica y petequias de las extremidades inferiores, que en la biopsia demuestran vasculitis leucocitoclástica. También ocurren nódulos cutáneos causados por granulomas necrosantes extravasculares y pápulas en los codos.3
HIPERLIPOPROTEINEMIA
Los xantomas son manifestaciones cutáneas de las hiperlipoproteinemias. Ocurren varios tipos de xantomas con diferentes alteraciones de lípidos. El xantelasma de los párpados es la manifestación más común de la hipercolesterolemia familiar; sin embargo, por lo menos la mitad de las personas que tienen lesiones en los párpados tienen lípidos normales en el plasma. Los xantomas planares son placas planas y amarillas que pueden afectar las palmas, plantas, cuello y tórax. Ocurren en pacientes con cirrosis biliar o mieloma múltiple. Los xantomas tuberosos son nódulos grandes amarillos o rojos que aparecen en las superficies extensoras de las articulaciones, como en los codos y las manos, pero no están fijos a los tendones subyacentes. Pueden ocurrir en pacientes con niveles elevados de triglicéridos o colesterol. Por el contrario, los xantomas tendinosos, que aparecen en pacientes con hipercolesterolemia familiar, están fijos a los tendones subyacentes de los codos, tobillos, rodillas y manos. Ocurren xantomas eruptivos cuando los niveles de triglicéridos en plasma se elevan en forma súbita. La lesiones cutáneas consisten en pápulas pequeñas y amarillas que con frecuencia se resuelven al disminuir los niveles de triglicéridos.
ENFERMEDAD DE KAWASAKI
La enfermedad de Kawasaki, también llamada síndrome ganglionar mucocutáneo, es un trastorno de la infancia que puede complicarse con oclusión arterial coronaria e infarto al miocardio, aneurismas de las arterias coronarias, alteraciones electrocardiográficas, arritmias cardiacas o miocarditis. Se ha sugerido que una toxina secretada por Staphylococcus aureus es responsable de esta enfermedad.4 El diagnóstico se basa en los criterios clínicos, que incluyen fiebre, conjuntivitis, linfadenopatía y erupción cutánea. Además de la erupción eritematosa generalizada, pueden desarrollarse alteraciones de la mucosa oral, así como edema y eritema de las manos y pies. Al final ocurre descamación importante de palmas y plantas. También son comunes el eritema perianal y escrotal y la cicatrización. La trombocitosis es un dato tardío, con cuentas de plaquetas que aumentan a más de un millón a las 2 semanas de inicio de la enfermedad.
SEUDOXANTOMA ELASTICO
El seudoxantoma elástico (SE) es un trastorno hereditario del tejido elástico que se asocia con muy diversas manifestaciones sistémicas. Las estrías angioides, la característica ocular del SE, son rupturas en la membrana de Bruch. Con frecuencia ocurren sangrado retiniano y pérdida de la visión. La calcificación de la lámina elástica interna de las arterias puede causar sangrado u oclusión de los vasos. En consecuencia, los pacientes desarrollan claudicación intermitente al caminar y enfermedad coronaria oclusiva a edad temprana. También se han descrito alteraciones valvulares cardiacas.5 Las lesiones cutáneas consisten en máculas amarillas semejantes a xantomas, pápulas o pliegues de piel redundantes en las áreas de flexión, en especial en el cuello y las axilas [ver figura 2]. Algunos pacientes pueden tener manifestaciones sistémicas del SE sin lesiones cutáneas clínicamente aparentes.6 El diagnóstico se establece por biopsia de la cicatriz o de la piel aparentemente normal de la zona de flexión.7 El defecto genético responsable del SE se ha mapeado en el cromosoma 16.8
FIEBRE REUMATICA
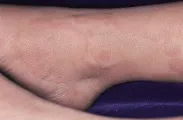
Las dos manifestaciones cutáneas de la fiebre reumática son el eritema marginado y los nódulos subcutáneos. El eritema marginado es una erupción eritematosa evanescente, anular y transitoria, que se desarrolla sobre las articulaciones [ver figura 3]. Los nódulos subcutáneos que aparecen en la fiebre reumática son no dolorosos, con movimiento libre y de alrededor de 1 cm de diámetro. Ocurren en las superficies extensoras de los codos, manos o pies.
SINDROME DE LAS UÑAS AMARILLAS
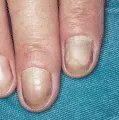
El síndrome de las uñas amarillas es causado por una alteración de los linfáticos [ver figura 4]. Los pacientes afectados desarrollan linfedema, por lo general de las piernas, y derrames pleurales. También son comunes los síntomas pulmonares, como la bronquitis recurrente. El diagnóstico se hace por la evidencia de función linfática anormal asociada con uñas amarillas y sin otras causas de patología ungueal.9
Enfermedades endócrinas
DIABETES MELLITUS
La diabetes mellitus se asocia con numerosas manifestaciones cutáneas . Puede ocurrir acantosis nigricans en pacientes con diabetes y otras endocrinopatías, como síndrome de Cushing, acromegalia, síndrome de ovarios poliquísticos y enfermedad tiroidea. Las lesiones cutáneas consisten en parches café aterciopelados en las zonas intertriginosas, en especial en cuello y axilas. La acantosis nigricans también se ha asociado con neoplasias internas, en especial adenocarcinoma gástrico u otros adenocarcinomas gastrointestinales.
La necrobiosis lipoídica es una manifestación cutánea específica de la diabetes. Las lesiones consisten en parches atróficos crónicos con bordes eritematosos que se expanden. Se afectan principalmente las piernas. Los centros de las lesiones son amarillos por la grasa subcutánea que es visible a través de la dermis atrófica y la epidermis. En ocasiones las lesiones se ulceran. La necrobiosis lipoídica suele asociarse con nefropatía diabética o retinopatía.10
El escleredema, otra manifestación de la diabetes, consiste en la induración de la piel de la espalda y parte posterior del cuello en pacientes obesos con diabetes tipo 2 (no insulino dependiente). El escleredema puede mejorar si se controla la diabetes.11 Con menos frecuencia ocurre escleredema en pacientes no diabéticos después de una faringitis estreptocócica, y en estos pacientes la enfermedad es autolimitada, resolviéndose a los 2 años de inicio.
En los pacientes diabéticos también ocurren bulas diabéticas, las úlceras neuropáticas y la llamada piel cérea y articulaciones rígidas. En esta última condición se desarrolla induración de la piel semejante a la de la esclerodermia en la piel sobre la cara dorsal de las manos, lo que impide la flexión o extensión completa de las articulaciones interfalángicas proximales.
Los pacientes diabéticos tienen propensión a diversas infecciones, incluyendo eritrasma, una infección por corinebacterias que provoca placas asintomáticas rojo-café en sitios intertriginosos, en especial en las ingles y axilas. También sufren infecciones por estafilococos y desarrollan forúnculos y carbuncos. La infección por Candida es otro riesgo, en especial cuando la glucemia está mal controlada.
ENFERMEDAD DE GRAVES
La enfermedad de Graves consiste en una triada de exoftalmos, hipertiroidismo y mixedema pretibial. El mixedema pretibial se manifiesta por nódulos del color de la piel y placas que se extienden desde el área pretibial hasta el dorso de los pies. Ocurre onicolisis, que es la separación de la placa de la uña del lecho ungueal, en muchos pacientes con hipertiroidismo. Otras enfermedades cutáneas autoinmunes, como el vitiligo y la alopecia areata, están aumentadas en los pacientes con enfermedad de Graves. Las lesiones suelen desarrollarse después del tratamiento del hipertiroidismo, aunque pueden ocurrir en cualquier fase de la evolución de la enfermedad de Graves. Otras manifestaciones de la enfermedad tiroidea incluyen los estigmas del hipotiroidismo. Los pacientes pueden desarrollar alopecia y pierden específicamente el tercio lateral de las cejas. También ocurre engrosamiento edematoso de los labios, lengua y nariz.
Enfermedades gastrointestinales
Los pacientes con diversas enfermedades gastrointestinales pueden presentar manifestaciones cutáneas. En forma semejante, los pacientes con ciertos padecimientos cutáneos pueden desarrollar complicaciones gastrointestinales.
SINDROME CARCINOIDE
El síndrome carcinoide se caracteriza por rubor episódico que puede asociarse con dolor abdominal, diarrea y sibilancias. El 90 por ciento de los tumores carcinoides se originan en el tubo digestivo; sin embargo, en ocasiones ocurren carcinoides bronquiales. Las manifestaciones cutáneas menos comunes de los tumores carcinoides incluyen cambios esclerodermatosos, metástasis cutáneas que se presentan como nódulos profundos e hiperqueratosis y pigmentación semejantes a las observadas en la pelagra.
ENFERMEDAD INFLAMATORIA DEL INTESTINO
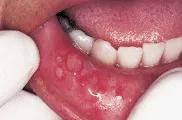
Existen varias manifestaciones cutáneas específicas e inespecíficas de la enfermedad inflamatoria del intestino. Tanto en la enfermedad de Crohn como en la colitis ulcerativa, los pacientes pueden progresar a un estado de hipercoagulabilidad, causando trombosis venosas y arteriales que pueden provocar pérdida de dedos y extremidades. La estomatitis aftosa es otra manifestación inespecífica de la enfermedad inflamatoria del intestino [ver figura 5]. En los pacientes con enfermedad de Crohn las lesiones pueden presentarse como granulomas no caseosos, mientras que en pacientes con colitis ulcerativa pueden ser indistinguibles de las úlceras neoplásicas.
Ocurre pioderma gangrenoso en los pacientes con enfermedad de Crohn y colitis ulcerativa, y también se ha reportado en enfermos con hepatitis activa crónica, artritis reumatoide y diversos trastornos mieloproliferativos. Las lesiones son indistinguibles de otras úlceras por la presencia de orificios semejantes a cráteres, pústulas y drenaje purulento [ver figura 6]. Pueden ocurrir en sitios de traumatismo. En ocasiones se necesita tratamiento con esteroides inyectados en forma intralesional o sistémicos. Se ha demostrado que los agentes inmunosupresores, como la ciclosporina, son muy eficaces. En los casos refractarios la talidomida ha demostrado ser benéfica.12
El eritema nodoso es una paniculitis septal que se asocia con diversas condiciones, incluyendo enfermedad de Crohn, colitis ulcerativa, síndrome de Behcet, sarcoidosis, infección y la ingestión de estrógenos y otros fármacos. Otras manifestaciones de la enfermedad de Crohn incluyen abscesos inguinales y fístulas sinusales y anales.
ENFERMEDAD DE CROHN METASTASICA
El término enfermedad de Crohn metastásica se refiere a la demostración demostrada por histología de granulomas no caseosos alejados del tubo digestivo en pacientes con enfermedad de Crohn. El cuadro clínico puede ser muy variable y el diagnóstico con frecuencia es erróneo. En algunos casos los pacientes presentan edema marcado del escroto o de la vulva.
TRASTORNOS CUTANEOS CON COMPLICACIONES GASTROINTESTINALES
Enfermedad de Cowden
La enfermedad de Cowden es un padecimiento autosómico dominante en el que se desarrollan pólipos gastrointestinales junto con numerosas lesiones de la piel. Ocurren pápulas parecidas a las verrugas conocidas como triquilemomas, en especial alrededor de la nariz, boca y oídos, pero también en las manos y pies [ver figura 7]. Pueden desarrollarse pápulas pequeñas sobre la mucosa gingival, creando una apariencia en empedrado. Son comunes los hemangiomas y los lipomas.13 Se ha descrito un nódulo característico del cuero cabelludo conocido como fibroma de Cowden. Hasta el 50 por ciento de las mujeres con enfermedad de Cowden desarrollan cáncer de mama. Ocurre también carcinoma, adenoma y bocio tiroideos.
Dermatitis herpetiforme
La dermatitis herpetiforme es una enfermedad vesicular autoinmune que se asocia con una enteropatía sensible a gluten. Las lesiones de la piel comienzan como vesículas que son tan pruriginosas que se rompen por el rascado, dejando solo excoriaciones y costras. Como los pacientes con enfermedad celiaca que no reciben dieta sin gluten, los pacientes con dermatitis herpetiforme tienen mayor riesgo de linfoma no Hodgkin.14
Síndrome de Peutz-Jeghers
En el síndrome de Peutz-Jeghers los pacientes desarrollan pólipos hamartomatosos del intestino delgado asociados con máculas pigmentadas de los labios y la mucosa oral. También pueden desarrollarse máculas en las palmas, plantas, dedos de manos y pies, y alrededor de la boca, la nariz y el recto. La enfermedad se hereda con un rasgo autosómico dominante y el defecto genético se ha localizado en el cromosoma 19.15
Epidermolisis bulosa distrófica recesiva
La epidermolisis bulosa distrófica recesiva es una enfermedad bulosa congénita con formación recurrente de ampollas y cicatrices, en especial en las manos y los pies. La cicatrización causa seudosindactilia, originando manos en mitones. La ingestión de alimentos burdos puede causar bulas mucosas en el esófago, que sanan dejando cicatrices y estenosis. La disfagia es una molestia frecuente. La cicatrización del esófago puede ocasionar carcinoma de células epidermoides, que es la principal causa de muerte en este padecimiento. Los gastroenterólogos y los dermatólogos tienen un papel importante en el manejo de estos pacientes. Las dietas líquidas y en puré, además del cuidado apropiado a la piel, son indispensables para la supervivencia de los pacientes que tienen este trastorno debilitante. El diagnóstico prenatal puede hacerse tomando muestras del ADN de las vellosidades coriónicas.16
Enfermedades hematológicas
AMILOIDOSIS
Existen varias formas de amiloidosis local y sistémica. En una forma asoicada con mieloma múltiple se depositan en la piel fibrillas de amiloide que consisten en cadenas ligeras de inmunoglobulina. Se desarrollan pápulas brillantes y translúcidas, en especial en los párpados. Debido al depósito de amiloide en los vasos sanguíneos ocurre sangrado espontáneo. Los traumatismos leves provocan petequias y púrpura. También ocurre macroglosia en algunos pacientes con amiloidosis asociada a mieloma y amiloidosis sistémica primaria. Las manifestaciones sistémicas de la amiloidosis asociada a mieloma y sistémica primaria son muy variadas. Ocurre hepatomegalia en el 50 por ciento de los pacientes. El amiloide puede afectar el corazón, causando insuficiencia cardiaca congestiva o infarto al miocardio. La amiloidosis del tubo digestivo puede causar malabsorción y enteropatía perdedora de proteínas.
MASTOCITOSIS
La mastocitosis es causada por infiltración de las células cebadas en la piel y otros órganos. La urticaria pigmentosa se refiere a las lesiones de la piel que ocurren en la mayoría de los pacientes con mastocitosis. Son características las máculas y pápulas rojo-café que recuerdan nevos [ver figura 8]. Al tallar las lesiones individuales ocurren ronchas urticarianas, un fenómeno conocido como signo de Darier. El prurito, el rubor, el dolor abdominal, la náusea, el vómito y la diarrea son molestias frecuentes.
La mayoría de los pacientes con mastocitosis tienen una forma indolente de enfermedad, incluso cuando los mastocitos infiltran la médula ósea.17 La enfermedad agresiva o maligna puede afectar el bazo, hígado y ganglios linfáticos, además de la piel y la médula osea. Desde el punto de vista histológico, los infiltrados contienen células cebadas atípicas no metacromáticas que son monoclonales en algunos pacientes.18
El diagnóstico de mastocitosis se hace por la demostración de células cebadas en la biopsia de piel. Debido a que las células cebadas se degranulan con facilidad, lo que hace difícil identificarlas, las biopsias deben realizarse con un mínimo de manipulación del tejido.
PORFIRIAS
Las porfirias se deben a una síntesis defectuosa de hemoglobina, causando porfirinas en exceso en la sangre y los tejidos corporales.
Porfiria eritropoyética congénita
La porfiria eritropoyética congénita es una enfermedad autosómica recesiva rara que se caracteriza por fotosensibilidad severa. Se desarrollan vesículas y bulas después de la exposición al sol, estas lesiones sanan con formación de cicatrices. Es común la pérdida de dedos, pabellones auriculares y la nariz en los pacientes que sobreviven hasta la edad adulta. La hipertricosis es otra complicación frecuente. La formación de cálculos vesiculares, la esplenomegalia y la anemia hemolítica se asocia también con este padecimiento.
Porfiria cutánea tarda
La porfiria cutánea tarda se caracteriza por fotosensibilidad, formación de vesículas (en especial en el dorso de las manos) e hipertricosis. La condición puede asociarse con ingestión de alcohol o medicamentos como estrógenos. El diagnóstico de porfiria puede establecerse por los niveles elevados de porfirina en la orina. El examen de la orina con lámpara de Wood revelará la fluorescencia rosa-roja atribuible al nivel aumentado de porfirinas urinarias.
Inmunodeficiencias
SIDA
La aparición del SIDA ha causado infecciones cutáneas y neoplasias que suelen tener una extensión y severidad dramáticas. Esta sección se describen algunas manifestaciones cutáneas de infecciones y otros padecimientos asociados con el SIDA.
Infecciones oportunistas
Infecciones virales Las infecciones virales banales, como el molusco contagioso, que ordinariamente se autolimitan y curan con facilidad, se vuelven diseminados, crónicos y graves en los pacientes con SIDA. Estas pápulas blancas umbilicadas, por lo general de solo algunos milímetros de diámetro, pueden alcanzar diámetros de 1 a 2 cm en los pacientes con SIDA. En forma semejante, el condiloma acuminado, causado por la infección por papilomavirus humano (PVH), suele ser difícil de tratar en los pacientes con SIDA.
Las infecciones por virus del herpes simple se vuelven crónicas y erosivas, formando úlceras grandes que no cicatrizan. Se han reportado cepas resistentes al aciclovir en algunos pacientes con SIDA;19 estos pacientes requieren otros agentes antivirales, como foscarnet. Se ha notificado que el cidovofir tópico (en gel) es benéfico para las infecciones por herpesvirus en pacientes infectados por VIH.20
Las infecciones por herpes zoster son un signo común de infección por VIH. En el huésped sin infección por VIH el herpes zoster se caracteriza por vesículas agrupadas en la distribución de un dermatoma. La erupción es autolimitada y se resuelve en 1 a 2 semanas. Por el contrario, la infección por herpes zoster puede causar una erupción vesicular diseminada en pacientes con SIDA, y en algunos se desarrollan lesiones crónicas que duran meses.
Infecciones micóticas Las infecciones micóticas son comunes en los pacientes con infección por VIH. Las infecciones por Candida incluyen el algodoncillo y la candidiasis inguinal. Varias infecciones micóticas severas que rara vez causan infección diseminada en pacientes con sistema inmunológico normal (v.gr., criptococosis, histoplasmosis, aspergilosis y esporotricosis) constituyen patógenos serios en los pacientes con SIDA.
Infecciones bacterianas Las infecciones bacterianas son más frecuentes y severas en pacientes con SIDA que en pacientes con sistema inmunológico normal. La angiomatosis bacilar, causada por Rochalimaea henselae, se presenta como pápulas y nódulos púrpura que pueden confundirse con sarcoma de Kaposi (ver adelante).21 Puede ocurrir fiebre crónica y calosfríos, lo mismo que lesiones óseas. La condición se resuelve al administrar antibióticos orales.
Escabiasis y otras erupciones pruriginosas La escabiasis, una erupción muy pruriginosa, tiene predilección por los glúteos, los genitales, el área periumbilical y los pliegues entre los dedos. Se ha descrito escabasis noruega, una forma psoriasiforme de costras gruesas, en pacientes con síndrome de Down y otros enfermos inmunosuprimidos, y en la actualidad se ve cada vez más en pacientes con SIDA. Las escamas de la escabiasis noruega contienen miles de ácaros que se ven con facilidad con el microscopio. Los surcos forman lesiones lineales de hasta 1 cm de largo. El ácaro causal, Sarcoptes scabiei, puede identificarse con el examen microscópico de los frotis de los surcos.
La foliculitis pustular eosinofílica y la erupción papular del SIDA son erupciones pruriginosas que afectan a los pacientes con infección por VIH. Ambas condiciones se caracterizan por prurito severo, pápulas del color de la piel y excoriaciones. Los pacientes con foliculitis pustular eosinofílica pueden desarrollar pústulas y pápulas eritematosas. Ambas condiciones responden al tratamiento con luz ultravioleta B.
Sarcoma de Kaposi
El sarcoma de Kaposi, una neoplasia vascular lentamente progresiva, se describió originalmente en varones ancianos italianos y judíos . Después se describió una forma más rápidamente progresiva en pacientes inmunosuprimidos con linfomas y pacientes con trasplante renal que recibían fármacos inmunosupresores. Se ha descrito una forma agresiva en pacientes con SIDA. El sarcoma de Kaposi clásico afecta típicamente las extremidades inferiores y progresa en forma gradual a otros sitios. Por el contrario, el sarcoma de Kaposi relacionado al SIDA puede ocurrir en cualquier superficie del organismo, incluyendo las mucosas. Se ha implicado al herpesvirus humano tipo 8 tanto en el sarcoma de Kaposi clásico como en el relacionado con el SIDA.22 Los tratamiento incluyen radioterapia, crioterapia e inyección intralesional con vinblastina.
Leucoplasia peluda oral
La leucoplasia peluda oral, otra condición que se ha descrito en pacientes infectados por VIH, consiste en pápulas blancas lineales sobre las superficies laterales de la lengua que causan la llamada apariencia peluda. La leucoplasia peluda oral puede distinguirse del algodoncillo oral porque las lesiones no pueden quitarse frotando, como en el algodoncillo.
OTROS ESTADOS DE INMUNODEFICIENCIA
Otros estados de inmunodeficiencia heredada o adquirida comparten diversas manifestaciones clínicas. La propensión a infecciones por monilia o infecciones bacterianas aumenta en trastornos como la enfermedad granulomatosa crónica y la neutropenia inducida por quimioterapia. También ocurren úlceras orales en la neutropenia cíclica y en la inmunosupresión inducida por quimioterapia.
Algunos medicamentos inmunosupresores tienen efectos cutáneos característicos. Los esteroides, cuando se usan por tiempo prolongado, causan fragilidad vascular, provocando púrpura esteroidea. También pueden provocar atrofia cutánea, formación de estrías y erupciones acneiformes. La ciclosporina se asocia con hipertricosis. La estomatitis aftosa es un efecto característico de numerosos medicamentos inmunosupresores, en especial de los agentes que disminuyen la función de la médula ósea.
Enfermedades infecciosas
Las manifestaciones cutáneas pueden ser importantes en diversas infecciones sistémicas, por ejemplo, los pacientes con septicemia pueden desarrollar coagulación intravascular diseminada (CID), que provoca hemorragias en la piel e infartos cutáneos. A continuación se describen características cutáneas clave de algunas infecciones sistémicas.
ENDOCARDITIS INFECCIOSA
Las manifestaciones cutáneas de la endocarditis infecciosa incluyen petequias, hemorragias en astilla (líneas rojas bajo la uña), nódulos de Osler (nódulos dolorosos y purpúricos en los cojinetes de los dedos de manos y pies) y lesiones de Janeway (máculas purpúricas no dolorosas de las palmas y plantas). Las lesiones cutáneas son causadas por embolias sépticas o vasculitis. El tratamiento de la infección subyacente causa resolución de las manifestaciones cutáneas.
SINDROME DE CHOQUE TOXICO
El síndrome de choque tóxico estafilocócico es una entidad descrita en forma reciente y que se detectó primero en mujeres menstruando que usaban tampones superabsorbentes. Es causado por una exotoxina producida por ciertas cepas de S. aureus.23 Se han implicado como causa las infecciones estafilocócicas en el hueso, tejidos blandos y otros sitios. Los pacientes desarrollan eritema difuso semejante al causado por quemadura solar con edema de las manos y pies, seguido de descamación de las palmas y plantas. También ocurre eritema de las mucosas, fiebre e hipotensión. Pueden desarrollarse síntomas gastrointestinales, menor función renal, elevación de las enzimas hepáticas, trombocitopenia y miositis.
SINDROME DE PIEL ESCALDADA POR ESTAFILOCOCO
El síndrome de piel escaldada por estafilococo (SSSS, por sus siglas en inglés, n. del t.) es causado por una toxina exfoliativa circulante producida por S. aureus fago grupo 11. La formación generalizada de bulas con grandes áreas de descamación es característica del padecimiento. Junto con el dolor, el eritema y la exfoliación de la piel, los pacientes tienen fiebre. No siempre es aparente el origen de la infección estafilocócica, y en ocasiones ésta se origina en una herida o un absceso oculto. Debido a que la infección estafilocócica suele estar lejos de la piel afectada, no crece S. aureus en el cultivo de piel.
El SSSS debe diferenciarse de la necrolisis epidérmica tóxica. Esta última afecta con frecuencia adultos y compromete las mucosas. El SSS suele afectar niños y respeta las mucosas. Además, la necrolisis epidérmica tóxica puede durar varias semanas y se asocia con una alta tasa de mortalidad, mientras que el SSSS dura pocos días y suele tener una buena evolución. Desde el punto de vista histológico, el SSSS muestra formación de bulas en la porción superior de la epidermis y la cavidad de la bula tiene células acantolíticas de apariencia normal y que flotan libres. En la necrolisis epidérmica tóxica ocurre formación de bulas en la capa basal de la epidermis y las células epidérmicas son necróticas.
FASCITIS NECROSANTE
La fascitis necrosante es causada por una infección anaeróbica mixta de una úlcera o una herida quirúrgica o traumática. La piel afectada está eritematosa, caliente y dolorosa, y desarrolla bulas hemorrágicas que se rompen para formar áreas de gangrena que crecen con rapidez y se extienden hacia abajo hasta la fascia. La debridación quirúrgica es indispensable para esta infección grave.24
MENINGOCOCCEMIA
La meningococcemia aguda puede ocurrir en epidemias o en casos aislados. Se desarrolla fiebre, cefalea y una erupción hemorrágica. Si no se trata los pacientes desarrollan CID con hemorragia importante, hipotensión y al final la muerte. El organismo causal, Neisseria meningititis, suele identificarse en el líquido cefalorraquídeo, pero también puede identificarse por frotis o cultivos de las lesiones de la piel o por hemocultivos.
FIEBRE ESCARLATINA
La fiebre escarlatina comienza con una faringitis causada por el estreptococo del grupo A. Se desarrolla una erupción generalizada 1 a 2 días después del inicio de la faringitis. La erupción se caracteriza por pápulas eritematosas puntiformes que es más fácil palpar que ver. Otras lesiones características incluyen una lengua en fresa y máculas petequiales lineales que ocurren en los pliegues del cuerpo (líneas de Pastia). Al disminuir la erupción aparece descamación de las palmas y las plantas. El tratamiento con penicilina causa resolución rápida de todos los síntomas.
INFECCION POR VIBRIO
La infección por Vibrio vulnificus se origina por traumatismos leves sufridos al nadar en lagos o el oceano o al limpiar alimentos del mar. Ocurre celulitis con linfangitis y bacteremia. En los pacientes con cirrosis hepática puede ocurrir infección después de comer ostras crudas. Estos pacientes desarrollan bulas hemorrágicas con leucopenia y coagulación intravascular diseminada.25
ENFERMEDAD DE LYME
La enfermedad de Lyme es causada por la espiroqueta Borrelia burgdorferi y se trasmite principalmente por la garrapata Ixodes dammini. La lesión cutánea característica, el eritema crónico migratorio, comienza como una mácula o una pápula eritematosa en el sitio de la mordedura de la garrapata. En días a semanas la lesión eritematosa se expande para formar un anillo rojo, con frecuencia con centro claro. Si no se tratan, las lesiones duran semanas o meses. Ocurre diseminación hematógena de las espiroquetas después de varias semanas, causando múltiples parches anulares de eritema crónico migratorio [ver figura 9]. Las complicaciones sistémicas incluyen una artritis aguda que afecta una o algunas articulaciones grandes pocas semanas después del inicio de los síntomas. Se desarrolla artritis erosiva crónica en alrededor del 10 por ciento de los pacientes. Pueden ocurrir síntomas neurológicos, incluyendo parálisis de Bell, lo mismo que complicaciones cardiacas, como insuficiencia cardiaca y alteraciones de la conducción.
La enfermedad de Lyme puede prevenirse eliminando las garrapatas en las primeras 18 horas después de que se fijan. Una vez que se desarrollan los síntomas los antibióticos orales son eficaces para destruir a B. burgdorferi. Una vacuna que contiene una proteína elaborada por ingeniería genética a partir de la superficie de la bacteria evita la infección en la mayoría de las personas vacunadas.26
FIEBRE MANCHADA DE LAS MONTAÑAS ROCOSAS
La fiebre manchada de las Montañas Rocosas (FMMR) es una enfermedad trasmitida por garrapatas causada por Rickettsia rickettsii. Se caracteriza por inicio súbito de fiebre, calosfríos y cefalea. Alrededor de 4 días después se desarrolla una erupción eritematosa característica que se vuelve purpúrica en las muñecas y tobillos. La erupción se disemina hacia el centro para afectar extremidades, tronco y cara.
Debido a que la mortalidad de la FMMR es alta, ante la sospecha clínica la enfermedad debe tratarse de inmediato con cloranfenicol intravenoso o tetraciclinas. El diagnóstico puede establecerse después por biopsia de piel: la inmunofluorescencia con anticuerpos contra R. rickettsii muestra el organismo en las paredes de los vasos sanguíneos cutáneos. Las pruebas serológicas, como la reacción de Weil-Felix, pueden confirmar el diagnóstico después de la fase aguda de la enfermedad.
Enfermedades neurológicas
SINDROME DE NEVOS DE CELULAS BASALES
El síndrome de nevos de células basales es un trastorno autosómico dominante en el que los pacientes desarrollan carcinomas de células basales en una etapa temprana. Con el síndrome se asocian múltiples alteraciones esqueléticas y los individuos afectados pueden desarrollar quistes en la mandíbula. Ocurre calcificación en lámina de la hoz del cerebro, así como otras alteraciones neurológicas, incluyendo meduloblastomas.
SINDROME DE NEVO EPIDERMICO
El síndrome de nevo epidérmico se caracteriza por manifestaciones sistémicas como crisis convulsivas, retraso mental, ceguera y alteraciones esqueléticas asociadas con nevos epidérmicos grandes. Los nevos consisten en tiras pigmentadas largas que son lineales o en remolino y que afectan grandes áreas del cuerpo [ver figura 10].
INCONTINENCIA DE PIGMENTO
La incontinencia de pigmento es un síndrome hereditario que afecta la piel y el sistema nervioso. El patrón de herencia es dominante ligado al X y es letal en fetos varones. La primera manifestación en la piel comienza a las semanas de nacimiento, en ocasiones in utero, y consiste en patrones lineales de lesiones vesiculobulosas. En semanas estas lesiones evolucionan a pápulas verrucosas y, eventualmente, a remolinos pigmentados. Aparte de los síntomas neurológicos, pueden ocurrir alteraciones oculares, alopecia cicatricial y malformaciones esqueléticas.
Hipomelanosis de Ito
La hipomelanosis de Ito, también llamada incontinencia de pigmento acromial, consiste en remolinos de hipopigmentación que se asocian con síntomas neurológicos en el 50 por ciento de los pacientes. Las lesiones cutáneas existen desde el nacimiento o se desarrollan en la infancia temprana. Además de las crisis convulsivas y el retraso mental, ocurren alteraciones esqueléticas y oculares.
NEUROFIBROMATOSIS
La neurofibromatosis es un padecimiento autosómico dominante común que afecta la piel y el sistema nervioso. Las lesiones cutáneas incluyen neurofibromas cutáneos, que son nódulos suaves, del color de la piel y con frecuencia pedunculados [ver figura 11]. Las manchas café con leche son planas, poco pigmentadas y hasta de varios centímetros de diámetro. Se encuentran seis o más manchas café con leche mayores de 1.5 cm de diámetro en la mayoría de los pacientes con neurofibromatosis tipo I (también llamada enfermedad de von Recklinhausen). Los neuromas plexiformes son tumores más grandes y profundos, que se asocian con hipertrofia ósea y de tejidos blandos. En una pequeña proporción de los tumores se originan neurofibrosarcomas. En la biopsia de piel las manchas café con leche contienen macromelanosomas, gránulos gigantes de pigmento en los melanocitos y los queratinocitos. También aparecen pecas axilares e inguinales como máculas pigmentadas que recuerdan manchas café con leche pequeñas en los sitios intertriginosos. Pueden encontrarse nódulos de Lisch (hamartomas pigmentados del iris) en la mayoría de los pacientes con neurofibromatosis.
Existen diferentes variantes de neurofibromatosis, incluyendo la neurofibromatosis segmentaria, en la que los pacientes desarrollan una distribución segmentaria de las manchas café con leche y neurofibromas cutáneos, y la neurofibromatosis tipo 2, que consiste en neuromas acústicos bilaterales sin nódulos de Lisch y con menos manchas café con leche que en el tipo 1. Los pacientes con neurofibromatosis tipo 2 pueden tener también algunos neurofibromas cutáneos. La neurofibromatosis tipos 1 y 2 son causadas por diferentes defectos genéticos, que se encuentran en los cromosomas 17 y 22, respectivamente.27,28
SINDROME DE SNEDDON
El síndrome de Sneddon es una enfermedad de la piel y del sistema nervioso causado por oclusión de arterias de tamaño pequeño a mediano en personas menores de 45 años. Las lesiones de la piel recuerdan al livedo reticularis y se han denominado livedo racemosa. Son frecuentes los episodios de isquemia transitoria o los eventos cerebrovasculares. El diagnóstico definitivo se realiza demostrando los cambios vasculares característicos en la biopsia de piel de los pacientes con datos neurológicos asociados.
ESCLEROSIS TUBEROSA
La esclerosis tuberosa es una enfermedad autosómico dominante que afecta la piel y el sistema nervioso central. Los pacientes afectados pueden desarrollar crisis convulsivas, retraso mental y lesiones cerebrales que pueden observarse en la TC. El adenoma sebáceo, la manifestación cutánea más característica de la esclerosis tuberosa, consiste en pápulas del color de la piel en la cara [ver figura 12a]. Otras lesiones son máculas hipopigmentadas denominadas manchas en hoja de fresno [ver figura 12b], lesiones hipopigmentadas más pequeñas conocidas como manchas en confeti, fibromas periungueales y subungueales (nódulos del color de la piel que se originan alrededor de las uñas de dedos de manos y pies) [ver figura 12c] y parches del color de la piel formados por tejido conectivo dérmico grueso. Se han identificado en los cromosomas 9 y 16 dos genes independientes que determinan la enfermedad.29
Enfermedades renales
ENFERMEDAD DE FABRY
La enfermedad de Fabry es causada por una alteración de la alfa-galactosidasa A, causando depósito de glucoesfingolípidos en los tejidos corporales. El trastorno se hereda con un rasgo recesivo ligado al X. Los varones afectados suelen quejarse de dolor severo en las extremidades, con ardor en las palmas y plantas. Los episodios de dolor son temporales, pero los pacientes refieren parestesias persistentes en manos y pies.
Las lesiones cutáneas consisten en angioqueratomas, que son pápulas rojas o purpúricas puntiformes que recuerdan hemangiomas de cereza [ver figura 13]. Los angioqueratomas se encuentran con más frecuencia en el área periumbilical, pero pueden ocurrir también en las palmas, plantas, tronco, extremidades y mucosas. En los adultos se depositan glucoesfingolípidos en los vasos sanguíneos y en los órganos, afectando el corazón, válvulas cardiacas, arterias coronarias y riñones.
POLIARTERITIS NODOSA
La poliarteritis nodosa es un trastorno inflamatorio que afecta las arterias musculares. Se forman aneurismas en muchas arterias, incluyendo las renales y del tejido subcutáneo. El diagnóstico de la forma sistémica de la poliarteritis puede hacerse demostrando aneurismas de las arterias renales en la angiografía renal.
La forma cutánea de poliarteritis nodosa se presenta principalmente como nódulos dolorosos en las extremidades inferiores.30 En los casos leves los pacientes pueden tener solo livedo reticularis, pero en los casos severos las lesiones cutáneas pueden ulcerarse. Puede existir polineuropatía asociada. Los pacientes con poliarteritis nodosa y microaneurismas tienen mayor incidencia de antigenemia de hepatitis B. A diferencia de otras vasculitis, suelen no tener anticuerpos anticitoplasma de neutrófilos.31
TRASTORNOS PERFORANTES
El término trastornos perforantes se refiere a varias condiciones que se caracterizan por salida de material dérmico a través de la epidermis. Estas lesiones se desarrollan con frecuencia asociadas con falla renal y diabetes mellitus.32 Las lesiones cutáneas se caracterizan por pápulas hiperqueratósicas con cráteres blancos centrales que histológicamente contienen material de la dermis. La colagenosis perforante reactiva, la foliculitis perforante y la enfermedad de Kyrle son todos ejemplos de trastornos perforantes asociados con insuficiencia renal.
CALCIFILAXIA
La calcifilaxia es un trastorno de pacientes con insuficiencia renal en la que áreas localizadas de la piel se vuelven necróticas por calcificación vascular. La calcifilaxia comienza con parches purpúricos dolorosos que pueden ser reticulados, recordando al livedo reticularis. Estos parches progresan a placas induradas que pueden ulcerarse, volviéndose necróticas [ver figura 14]. La calcifilaxia suele terminar en amputación o la muerte. La paratiroidectomía puede causar cicatrización de la piel afectada sin amputación.33
Enfermedades reumatológicas
DERMATOMIOSITIS
Las manifestaciones cutáneas más conocidas de la dermatomiositis, un trastorno inflamatorio del músculo y la piel, son las pápulas de Gottron y el eritema en heliotropo. Las pápulas de Gottron son máculas y pápulas descamativas eritematosas que ocurren en el dorso de los nudillos [ver figura 15]. El eritema en heliotropo consiste en eritema y edema periorbitario. Recientemente se han descrito lesiones en el cuero cabelludo, que pueden asociarse con alopecia.34 Las lesiones con frecuencia se confunden con dermatitis seborreica o psoriasis.
La asociación entre dermatomiositis y neoplasias sigue siento motivo de controversia. Se reportan neoplasias en el 15 a 50 por ciento de los pacientes, que suelen detectarse en la historia clínica y examen físico o en las pruebas de rutina, como mamografía, radiografía de tórax, examen de orina y examen de excremento para sangre oculta en heces. Rara vez se justifica la realización de más estudios, a menos que se encuentre evidencia de neoplasia en la evaluación inicial.35
Las clasificaciones de la dermatomiositis incluyen una variante juvenil caracterizada por calcificación de la piel o del músculo. La forma vasculítica en niños se complica por infartos cutáneos y ulceración, y por vasculitis gastrointestinal con dolor abdominal, hemorragia o perforación. Esta forma con vasculitis se asocia con mal pronóstico, y muchos de los pacientes fallecen por la enfermedad.
ESCLERODERMIA Y ENFERMEDADES SEMEJANTES
La esclerodermia incluye diversos síndromes diferentes que comparten una característica en común, la induración de la piel.
Esclerosis sistémica progresiva
La esclerosis sistémica progresiva, también conocida como esclerodermia sistémica, es una enfermedad frecuentemente fatal en la que los pacientes presentan fenómeno de Raynaud y esclerodactilia (induración de la piel de los dedos) [ver figura 16]. La induración cutánea puede volverse generalizada. La afección de la cara provoca una apariencia característica con fruncimiento de los labios y acartonamiento de la piel bajo la nariz que da la apariencia de pico de pájaro. Los pacientes con anticuerpos contra Scl-70 tienen mal pronóstico, falleciendo por enfermedad renal e hipertensión maligna. Los pacientes con anticuerpos anticentrómero tienen una variante de esclerodermia más lentamente progresiva conocida como síndrome de CREST, que se caracteriza por calcinosis cutánea, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia y telangiectasias. Con el tiempo se desarrolla fibrosis pulmonar, hipertensión pulmonar y falla cardiaca derecha.
Morfea
La morfea, también denominada esclerodermia localizada, se caracteriza por parches bien delimitados de piel indurada que pueden volverse generalizados. Se distingue de la esclerosis sistémica progresiva por al ausencia de fenómeno de Raynaud, esclerodactilia o complicaciones sistémicas de la esclerodermia.
Existen innovaciones en el tratamiento tanto de la esclerosis sistémica progresiva como de la morfea. Se ha reportado que la exposición a psoralenos y luz ultravioleta de onda larga (PUVA) mejora la esclerosis sistémica progresiva y la morfea en forma dramática,36 y la exposición a UVA1 (el espectro de UVA más largo, de 340 a 400 nm) beneficia la esclerodermia localizada.37 Deben hacerse más estudios para confirmar la eficacia de estos tratamientos. Estudios anecdóticos han indicado que la minociclina puede ser eficaz en la esclerosis sistémica progresiva, pero se requieren estudios controlados al respecto.38
Enfermedad injerto contra huésped
Al volverse más común el trasplante de órganos ha aumentado en frecuencia otra enfermedad parecida a la esclerodermia, la enfermedad injerto contra huésped, en especial después del trasplante de médula ósea. Existen dos fases en la enfermedad injerto contra huésped. La primera, la fase aguda, se desarrolla 10 a 40 días después del trasplante y consiste en una erupción macular y papular eritematosa que suele asociarse con fiebre, hepatomegalia, linfadenopatía o síntomas gastrointestinales. La enfermedad injerto contra huésped crónica se desarrolla 3 meses después del trasplante y consiste en pápulas purpúricas que recuerdan liquen plano [ver figura 17]. Lo más característico son los cambios cutáneos esclerodermatosos con telangiectasias, hiperpigmentación reticulada y alopecia. Tanto la ciclosporina como la PUVA han demostrado ser útiles en la prevención y tratamiento de la enfermedad injerto contra huésped.39,40
Fascitis eosinofílica
Ocurre endurecimiento de la piel parecido a la esclerodermia en la fascitis eosinofílica. Típicamente se desarrollan pliegues de la piel de las extremidades asociados con dolor. A diferencia de la esclerosis sistémica progresiva, no ocurre fenómeno de Raynaud. El diagnóstico definitivo requiere biopsia de la piel y la fascia suprayacente al músculo afectado. En algunos casos de fascitis eosinofílica se desarrollan alteraciones hematológicas, incluyendo anemia aplástica, trombocitopenia, enfermedad de Hodgkin y leucemias.41
Síndrome de mialgia eosinofilia
Algunos pacientes con diagnóstico erróneo de fascitis eosinofílica habían ingerido L-triptofano, que se ha asociado con el síndrome de mialgia eosinofilia. Como resultado ocurre dolor y debilidad musculares severas, junto con fiebre, eosinofilia y diversos síntomas sistémicos. Las manifestaciones cutáneas más comunes son eritema y edema.42
LUPUS ERITEMATOSO GENERALIZADO
Existen muchas manifestaciones cutáneas en el lupus, incluyendo fenómeno de Raynaud, el llamado lupus discoide (caracterizado por lesiones cicatriciales redondas con hipopigmentación central y un borde de hiperpigmentación), eritema malar, fotosensibilidad, alopecia y úlceras mucosas. Al aprender más sobre lupus el espectro de las enfermedades cutáneas asociadas con este trastorno continúa expandiéndose. El lupus cutáneo subagudo, una variante caracterizada serológicamente por anticuerpos anti-Ro y anti-La, se asocia con lesiones psoriasiformes anulares de la piel [ver figura 18].
Síndrome de anticuerpos anticardiolipina
El síndrome de anticuerpos anticardiolipina, que puede ocurrir en los pacientes con lupus, se ha descrito en pacientes que sufren episodios repetidos de flebitis, trombosis arteriales y abortos de repetición. Son comunes los infartos cutáneos. Los pacientes tienen serología falsa positiva para sífilis y tienen anticoagulante lúpico circulante. Los anticuerpos antifosfolípido circulantes son la característica serológica de este síndrome recién descrito.43
Livedo vasculitis
La livedo vasculitis, otra entidad que se ha asociado con el lupus, se caracteriza por úlceras dolorosas y recurrentes en las piernas y tobillos. Las úlceras sanan dejando cicatrices escleróticas blancas. Los pacientes afectados con frecuencia tienen livedo reticularis. Esta condición, también conocida como atrofia blanca, se ha atribuido a procesos trombóticos más que a depósito de complejos inmunes o vasculitis leucocitoclástica.44
Lupus neonatal
El lupus neonatal es un síndrome caracterizado por máculas y pápulas eritematosas anulares que ocurren en la cara de niños recién nadidos. El trastorno se ha atribuido al paso transplacentario de anticuerpos anti-Ro y en ocasiones anti-La. Las madres con frecuencia están asintomáticas, pero pueden tener lupus o síndrome de Sjögren. El bloqueo cardiaco congénito es la complicación más seria de este padecimiento.
DR. MARK LEBWOHL
Las manifestaciones cutáneas de las enfermedades sistémicas son tan numerosas y diversas que es imposible abarcarlas en un solo capítulo, incluso en forma somera. En lugar de ello, este capítulo revisa manifestaciones cutáneas claves de enfermedades sistémicas que deben ser detectadas por la mayoría de los médicos y destaca los adelantos recientes en el diagnóstico y tratamiento de estos padecimientos. Para mayor descripción de las enfermedades específicas, incluyendo sus manifestaciones cutáneas, los lectores deben consultar los capítulos dedicados a estos padecimientos.
En muchos de los trastornos presentados en este capítulo, la evaluación y tratamiento del padecimiento sistémico subyacente son indispensables para tener una evolución favorable. Por ejemplo, el dato de sarcoidosis cutánea obliga a buscar sarcoidosis sistémica. En otros padecimientos, por ejemplo en la epidermolisis bulosa distrófica recesiva, el tratamiento del trastorno cutáneo es clave para el manejo de la enfermedad sistémica.
Enfermedades cardiopulmonares y vasculares
SARCOIDOSIS
Las manifestaciones cutáneas de la sarcoidosis son tan variadas como sus manifestaciones sistémicas. Las pápulas alrededor de los ojos y la nariz son las más características. El término lupus pernio se refiere a granulomas no caseosos que provocan placas translúcidas y violáceas en los oídos, mejillas y nariz [ver figura 1]. Puede ocurrir afección del hueso subyacente y el diagnóstico se realiza por biopsia de piel. El tratamiento tradicional consiste en esteroides intralesionales y se han usado con éxito antipalúdicos y metotrexate. En algunos pacientes con sarcoidosis ocurre eritema nodoso, caracterizado por nódulos profundos, dolorosos y eritematosos en las extremidades inferiores. El lupus pernio se asocia con una evolución más crónica, mientras que el eritema nodoso indica una enfermedad aguda y más benigna.1
|
| Figura 1 |
| Sarcoidosis |
VASCULITIS GRANULOMATOSA
Granulomatosis de Wegener
La granulomatosis de Wegener se asocia con signos mucocutáneos tanto distintivos como no específicos. La púrpura palpable es uno de los datos cutáneos más comunes, aunque también se han descrito úlceras, pápulas, nódulos y bulas. Además de los síntomas de vías respiratorias superiores y pulmonares , la presencia de deformidad de la nariz en silla de montar, ulceraciones nasales y perforación septal debe sugerir el diagnóstico de granulomatosis de Wegener. El diagnóstico definitivo se realiza demostrando vasculitis con granulomas no caseosos en un paciente con afección de vías respiratorias superiores e inferiores y glomerulonefritis. Con frecuencia existen anticuerpos anticitoplasma de neutrófilo.
Granulomatosis linfomatoide
La granulomatosis linfomatoide puede asociarse con lesiones cutáneas inespecíficas, incluyendo máculas eritematosas, pápulas, placas, nódulos y úlceras. Este trastorno se distingue clínicamente de la granulomatosis de Wegener por la ausencia de afección de las vías respiratorias superiores. El diagnóstico se establece al demostrar un infiltrado necrosante granulomatoso con células linfoides atípicas alrededor de los vasos sanguíneos. Este trastorno se ha atribuido a proliferación clonal de las células B que induce una respuesta de células T.2
Síndrome de Churg-Strauss
El síndrome de Churg-Strauss, o angeítis granulomatosa alérgica, se presenta con más frecuencia como asma y eosinofilia; sin embargo, se desarrollan lesiones cutáneas en hasta el 40 por ciento de los pacientes. Los datos más comunes consisten en púrpura palpable simétrica y petequias de las extremidades inferiores, que en la biopsia demuestran vasculitis leucocitoclástica. También ocurren nódulos cutáneos causados por granulomas necrosantes extravasculares y pápulas en los codos.3
HIPERLIPOPROTEINEMIA
Los xantomas son manifestaciones cutáneas de las hiperlipoproteinemias. Ocurren varios tipos de xantomas con diferentes alteraciones de lípidos. El xantelasma de los párpados es la manifestación más común de la hipercolesterolemia familiar; sin embargo, por lo menos la mitad de las personas que tienen lesiones en los párpados tienen lípidos normales en el plasma. Los xantomas planares son placas planas y amarillas que pueden afectar las palmas, plantas, cuello y tórax. Ocurren en pacientes con cirrosis biliar o mieloma múltiple. Los xantomas tuberosos son nódulos grandes amarillos o rojos que aparecen en las superficies extensoras de las articulaciones, como en los codos y las manos, pero no están fijos a los tendones subyacentes. Pueden ocurrir en pacientes con niveles elevados de triglicéridos o colesterol. Por el contrario, los xantomas tendinosos, que aparecen en pacientes con hipercolesterolemia familiar, están fijos a los tendones subyacentes de los codos, tobillos, rodillas y manos. Ocurren xantomas eruptivos cuando los niveles de triglicéridos en plasma se elevan en forma súbita. La lesiones cutáneas consisten en pápulas pequeñas y amarillas que con frecuencia se resuelven al disminuir los niveles de triglicéridos.
ENFERMEDAD DE KAWASAKI
La enfermedad de Kawasaki, también llamada síndrome ganglionar mucocutáneo, es un trastorno de la infancia que puede complicarse con oclusión arterial coronaria e infarto al miocardio, aneurismas de las arterias coronarias, alteraciones electrocardiográficas, arritmias cardiacas o miocarditis. Se ha sugerido que una toxina secretada por Staphylococcus aureus es responsable de esta enfermedad.4 El diagnóstico se basa en los criterios clínicos, que incluyen fiebre, conjuntivitis, linfadenopatía y erupción cutánea. Además de la erupción eritematosa generalizada, pueden desarrollarse alteraciones de la mucosa oral, así como edema y eritema de las manos y pies. Al final ocurre descamación importante de palmas y plantas. También son comunes el eritema perianal y escrotal y la cicatrización. La trombocitosis es un dato tardío, con cuentas de plaquetas que aumentan a más de un millón a las 2 semanas de inicio de la enfermedad.
SEUDOXANTOMA ELASTICO
El seudoxantoma elástico (SE) es un trastorno hereditario del tejido elástico que se asocia con muy diversas manifestaciones sistémicas. Las estrías angioides, la característica ocular del SE, son rupturas en la membrana de Bruch. Con frecuencia ocurren sangrado retiniano y pérdida de la visión. La calcificación de la lámina elástica interna de las arterias puede causar sangrado u oclusión de los vasos. En consecuencia, los pacientes desarrollan claudicación intermitente al caminar y enfermedad coronaria oclusiva a edad temprana. También se han descrito alteraciones valvulares cardiacas.5 Las lesiones cutáneas consisten en máculas amarillas semejantes a xantomas, pápulas o pliegues de piel redundantes en las áreas de flexión, en especial en el cuello y las axilas [ver figura 2]. Algunos pacientes pueden tener manifestaciones sistémicas del SE sin lesiones cutáneas clínicamente aparentes.6 El diagnóstico se establece por biopsia de la cicatriz o de la piel aparentemente normal de la zona de flexión.7 El defecto genético responsable del SE se ha mapeado en el cromosoma 16.8

|
| Figura 2 |
| Seudoxantoma elástico |
FIEBRE REUMATICA
Las dos manifestaciones cutáneas de la fiebre reumática son el eritema marginado y los nódulos subcutáneos. El eritema marginado es una erupción eritematosa evanescente, anular y transitoria, que se desarrolla sobre las articulaciones [ver figura 3]. Los nódulos subcutáneos que aparecen en la fiebre reumática son no dolorosos, con movimiento libre y de alrededor de 1 cm de diámetro. Ocurren en las superficies extensoras de los codos, manos o pies.
|
| Figura 3 |
| Fiebre reumática |
SINDROME DE LAS UÑAS AMARILLAS
El síndrome de las uñas amarillas es causado por una alteración de los linfáticos [ver figura 4]. Los pacientes afectados desarrollan linfedema, por lo general de las piernas, y derrames pleurales. También son comunes los síntomas pulmonares, como la bronquitis recurrente. El diagnóstico se hace por la evidencia de función linfática anormal asociada con uñas amarillas y sin otras causas de patología ungueal.9
|
| Figura 4 |
| Síndrome de uñas amarillas |
Enfermedades endócrinas
DIABETES MELLITUS
La diabetes mellitus se asocia con numerosas manifestaciones cutáneas . Puede ocurrir acantosis nigricans en pacientes con diabetes y otras endocrinopatías, como síndrome de Cushing, acromegalia, síndrome de ovarios poliquísticos y enfermedad tiroidea. Las lesiones cutáneas consisten en parches café aterciopelados en las zonas intertriginosas, en especial en cuello y axilas. La acantosis nigricans también se ha asociado con neoplasias internas, en especial adenocarcinoma gástrico u otros adenocarcinomas gastrointestinales.
La necrobiosis lipoídica es una manifestación cutánea específica de la diabetes. Las lesiones consisten en parches atróficos crónicos con bordes eritematosos que se expanden. Se afectan principalmente las piernas. Los centros de las lesiones son amarillos por la grasa subcutánea que es visible a través de la dermis atrófica y la epidermis. En ocasiones las lesiones se ulceran. La necrobiosis lipoídica suele asociarse con nefropatía diabética o retinopatía.10
El escleredema, otra manifestación de la diabetes, consiste en la induración de la piel de la espalda y parte posterior del cuello en pacientes obesos con diabetes tipo 2 (no insulino dependiente). El escleredema puede mejorar si se controla la diabetes.11 Con menos frecuencia ocurre escleredema en pacientes no diabéticos después de una faringitis estreptocócica, y en estos pacientes la enfermedad es autolimitada, resolviéndose a los 2 años de inicio.
En los pacientes diabéticos también ocurren bulas diabéticas, las úlceras neuropáticas y la llamada piel cérea y articulaciones rígidas. En esta última condición se desarrolla induración de la piel semejante a la de la esclerodermia en la piel sobre la cara dorsal de las manos, lo que impide la flexión o extensión completa de las articulaciones interfalángicas proximales.
Los pacientes diabéticos tienen propensión a diversas infecciones, incluyendo eritrasma, una infección por corinebacterias que provoca placas asintomáticas rojo-café en sitios intertriginosos, en especial en las ingles y axilas. También sufren infecciones por estafilococos y desarrollan forúnculos y carbuncos. La infección por Candida es otro riesgo, en especial cuando la glucemia está mal controlada.
ENFERMEDAD DE GRAVES
La enfermedad de Graves consiste en una triada de exoftalmos, hipertiroidismo y mixedema pretibial. El mixedema pretibial se manifiesta por nódulos del color de la piel y placas que se extienden desde el área pretibial hasta el dorso de los pies. Ocurre onicolisis, que es la separación de la placa de la uña del lecho ungueal, en muchos pacientes con hipertiroidismo. Otras enfermedades cutáneas autoinmunes, como el vitiligo y la alopecia areata, están aumentadas en los pacientes con enfermedad de Graves. Las lesiones suelen desarrollarse después del tratamiento del hipertiroidismo, aunque pueden ocurrir en cualquier fase de la evolución de la enfermedad de Graves. Otras manifestaciones de la enfermedad tiroidea incluyen los estigmas del hipotiroidismo. Los pacientes pueden desarrollar alopecia y pierden específicamente el tercio lateral de las cejas. También ocurre engrosamiento edematoso de los labios, lengua y nariz.
Enfermedades gastrointestinales
Los pacientes con diversas enfermedades gastrointestinales pueden presentar manifestaciones cutáneas. En forma semejante, los pacientes con ciertos padecimientos cutáneos pueden desarrollar complicaciones gastrointestinales.
SINDROME CARCINOIDE
El síndrome carcinoide se caracteriza por rubor episódico que puede asociarse con dolor abdominal, diarrea y sibilancias. El 90 por ciento de los tumores carcinoides se originan en el tubo digestivo; sin embargo, en ocasiones ocurren carcinoides bronquiales. Las manifestaciones cutáneas menos comunes de los tumores carcinoides incluyen cambios esclerodermatosos, metástasis cutáneas que se presentan como nódulos profundos e hiperqueratosis y pigmentación semejantes a las observadas en la pelagra.
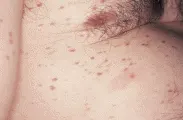
ENFERMEDAD INFLAMATORIA DEL INTESTINO
Existen varias manifestaciones cutáneas específicas e inespecíficas de la enfermedad inflamatoria del intestino. Tanto en la enfermedad de Crohn como en la colitis ulcerativa, los pacientes pueden progresar a un estado de hipercoagulabilidad, causando trombosis venosas y arteriales que pueden provocar pérdida de dedos y extremidades. La estomatitis aftosa es otra manifestación inespecífica de la enfermedad inflamatoria del intestino [ver figura 5]. En los pacientes con enfermedad de Crohn las lesiones pueden presentarse como granulomas no caseosos, mientras que en pacientes con colitis ulcerativa pueden ser indistinguibles de las úlceras neoplásicas.
|
| Figura 5 |
| Estomatitis aftosa |
Ocurre pioderma gangrenoso en los pacientes con enfermedad de Crohn y colitis ulcerativa, y también se ha reportado en enfermos con hepatitis activa crónica, artritis reumatoide y diversos trastornos mieloproliferativos. Las lesiones son indistinguibles de otras úlceras por la presencia de orificios semejantes a cráteres, pústulas y drenaje purulento [ver figura 6]. Pueden ocurrir en sitios de traumatismo. En ocasiones se necesita tratamiento con esteroides inyectados en forma intralesional o sistémicos. Se ha demostrado que los agentes inmunosupresores, como la ciclosporina, son muy eficaces. En los casos refractarios la talidomida ha demostrado ser benéfica.12
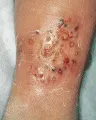
|
| Figura 6 |
| Pioderma gangrenoso |
El eritema nodoso es una paniculitis septal que se asocia con diversas condiciones, incluyendo enfermedad de Crohn, colitis ulcerativa, síndrome de Behcet, sarcoidosis, infección y la ingestión de estrógenos y otros fármacos. Otras manifestaciones de la enfermedad de Crohn incluyen abscesos inguinales y fístulas sinusales y anales.
ENFERMEDAD DE CROHN METASTASICA
El término enfermedad de Crohn metastásica se refiere a la demostración demostrada por histología de granulomas no caseosos alejados del tubo digestivo en pacientes con enfermedad de Crohn. El cuadro clínico puede ser muy variable y el diagnóstico con frecuencia es erróneo. En algunos casos los pacientes presentan edema marcado del escroto o de la vulva.
TRASTORNOS CUTANEOS CON COMPLICACIONES GASTROINTESTINALES
Enfermedad de Cowden
La enfermedad de Cowden es un padecimiento autosómico dominante en el que se desarrollan pólipos gastrointestinales junto con numerosas lesiones de la piel. Ocurren pápulas parecidas a las verrugas conocidas como triquilemomas, en especial alrededor de la nariz, boca y oídos, pero también en las manos y pies [ver figura 7]. Pueden desarrollarse pápulas pequeñas sobre la mucosa gingival, creando una apariencia en empedrado. Son comunes los hemangiomas y los lipomas.13 Se ha descrito un nódulo característico del cuero cabelludo conocido como fibroma de Cowden. Hasta el 50 por ciento de las mujeres con enfermedad de Cowden desarrollan cáncer de mama. Ocurre también carcinoma, adenoma y bocio tiroideos.

|
| Figura 7 |
| Enfermedad de Cowden |
Dermatitis herpetiforme
La dermatitis herpetiforme es una enfermedad vesicular autoinmune que se asocia con una enteropatía sensible a gluten. Las lesiones de la piel comienzan como vesículas que son tan pruriginosas que se rompen por el rascado, dejando solo excoriaciones y costras. Como los pacientes con enfermedad celiaca que no reciben dieta sin gluten, los pacientes con dermatitis herpetiforme tienen mayor riesgo de linfoma no Hodgkin.14
Síndrome de Peutz-Jeghers
En el síndrome de Peutz-Jeghers los pacientes desarrollan pólipos hamartomatosos del intestino delgado asociados con máculas pigmentadas de los labios y la mucosa oral. También pueden desarrollarse máculas en las palmas, plantas, dedos de manos y pies, y alrededor de la boca, la nariz y el recto. La enfermedad se hereda con un rasgo autosómico dominante y el defecto genético se ha localizado en el cromosoma 19.15
Epidermolisis bulosa distrófica recesiva
La epidermolisis bulosa distrófica recesiva es una enfermedad bulosa congénita con formación recurrente de ampollas y cicatrices, en especial en las manos y los pies. La cicatrización causa seudosindactilia, originando manos en mitones. La ingestión de alimentos burdos puede causar bulas mucosas en el esófago, que sanan dejando cicatrices y estenosis. La disfagia es una molestia frecuente. La cicatrización del esófago puede ocasionar carcinoma de células epidermoides, que es la principal causa de muerte en este padecimiento. Los gastroenterólogos y los dermatólogos tienen un papel importante en el manejo de estos pacientes. Las dietas líquidas y en puré, además del cuidado apropiado a la piel, son indispensables para la supervivencia de los pacientes que tienen este trastorno debilitante. El diagnóstico prenatal puede hacerse tomando muestras del ADN de las vellosidades coriónicas.16
Enfermedades hematológicas
AMILOIDOSIS
Existen varias formas de amiloidosis local y sistémica. En una forma asoicada con mieloma múltiple se depositan en la piel fibrillas de amiloide que consisten en cadenas ligeras de inmunoglobulina. Se desarrollan pápulas brillantes y translúcidas, en especial en los párpados. Debido al depósito de amiloide en los vasos sanguíneos ocurre sangrado espontáneo. Los traumatismos leves provocan petequias y púrpura. También ocurre macroglosia en algunos pacientes con amiloidosis asociada a mieloma y amiloidosis sistémica primaria. Las manifestaciones sistémicas de la amiloidosis asociada a mieloma y sistémica primaria son muy variadas. Ocurre hepatomegalia en el 50 por ciento de los pacientes. El amiloide puede afectar el corazón, causando insuficiencia cardiaca congestiva o infarto al miocardio. La amiloidosis del tubo digestivo puede causar malabsorción y enteropatía perdedora de proteínas.
MASTOCITOSIS
La mastocitosis es causada por infiltración de las células cebadas en la piel y otros órganos. La urticaria pigmentosa se refiere a las lesiones de la piel que ocurren en la mayoría de los pacientes con mastocitosis. Son características las máculas y pápulas rojo-café que recuerdan nevos [ver figura 8]. Al tallar las lesiones individuales ocurren ronchas urticarianas, un fenómeno conocido como signo de Darier. El prurito, el rubor, el dolor abdominal, la náusea, el vómito y la diarrea son molestias frecuentes.
|
| Figura 8 |
| Urticaria pigmentosa |
La mayoría de los pacientes con mastocitosis tienen una forma indolente de enfermedad, incluso cuando los mastocitos infiltran la médula ósea.17 La enfermedad agresiva o maligna puede afectar el bazo, hígado y ganglios linfáticos, además de la piel y la médula osea. Desde el punto de vista histológico, los infiltrados contienen células cebadas atípicas no metacromáticas que son monoclonales en algunos pacientes.18
El diagnóstico de mastocitosis se hace por la demostración de células cebadas en la biopsia de piel. Debido a que las células cebadas se degranulan con facilidad, lo que hace difícil identificarlas, las biopsias deben realizarse con un mínimo de manipulación del tejido.
PORFIRIAS
Las porfirias se deben a una síntesis defectuosa de hemoglobina, causando porfirinas en exceso en la sangre y los tejidos corporales.
Porfiria eritropoyética congénita
La porfiria eritropoyética congénita es una enfermedad autosómica recesiva rara que se caracteriza por fotosensibilidad severa. Se desarrollan vesículas y bulas después de la exposición al sol, estas lesiones sanan con formación de cicatrices. Es común la pérdida de dedos, pabellones auriculares y la nariz en los pacientes que sobreviven hasta la edad adulta. La hipertricosis es otra complicación frecuente. La formación de cálculos vesiculares, la esplenomegalia y la anemia hemolítica se asocia también con este padecimiento.
Porfiria cutánea tarda
La porfiria cutánea tarda se caracteriza por fotosensibilidad, formación de vesículas (en especial en el dorso de las manos) e hipertricosis. La condición puede asociarse con ingestión de alcohol o medicamentos como estrógenos. El diagnóstico de porfiria puede establecerse por los niveles elevados de porfirina en la orina. El examen de la orina con lámpara de Wood revelará la fluorescencia rosa-roja atribuible al nivel aumentado de porfirinas urinarias.
Inmunodeficiencias
SIDA
La aparición del SIDA ha causado infecciones cutáneas y neoplasias que suelen tener una extensión y severidad dramáticas. Esta sección se describen algunas manifestaciones cutáneas de infecciones y otros padecimientos asociados con el SIDA.
Infecciones oportunistas
Infecciones virales Las infecciones virales banales, como el molusco contagioso, que ordinariamente se autolimitan y curan con facilidad, se vuelven diseminados, crónicos y graves en los pacientes con SIDA. Estas pápulas blancas umbilicadas, por lo general de solo algunos milímetros de diámetro, pueden alcanzar diámetros de 1 a 2 cm en los pacientes con SIDA. En forma semejante, el condiloma acuminado, causado por la infección por papilomavirus humano (PVH), suele ser difícil de tratar en los pacientes con SIDA.
Las infecciones por virus del herpes simple se vuelven crónicas y erosivas, formando úlceras grandes que no cicatrizan. Se han reportado cepas resistentes al aciclovir en algunos pacientes con SIDA;19 estos pacientes requieren otros agentes antivirales, como foscarnet. Se ha notificado que el cidovofir tópico (en gel) es benéfico para las infecciones por herpesvirus en pacientes infectados por VIH.20
Las infecciones por herpes zoster son un signo común de infección por VIH. En el huésped sin infección por VIH el herpes zoster se caracteriza por vesículas agrupadas en la distribución de un dermatoma. La erupción es autolimitada y se resuelve en 1 a 2 semanas. Por el contrario, la infección por herpes zoster puede causar una erupción vesicular diseminada en pacientes con SIDA, y en algunos se desarrollan lesiones crónicas que duran meses.
Infecciones micóticas Las infecciones micóticas son comunes en los pacientes con infección por VIH. Las infecciones por Candida incluyen el algodoncillo y la candidiasis inguinal. Varias infecciones micóticas severas que rara vez causan infección diseminada en pacientes con sistema inmunológico normal (v.gr., criptococosis, histoplasmosis, aspergilosis y esporotricosis) constituyen patógenos serios en los pacientes con SIDA.
Infecciones bacterianas Las infecciones bacterianas son más frecuentes y severas en pacientes con SIDA que en pacientes con sistema inmunológico normal. La angiomatosis bacilar, causada por Rochalimaea henselae, se presenta como pápulas y nódulos púrpura que pueden confundirse con sarcoma de Kaposi (ver adelante).21 Puede ocurrir fiebre crónica y calosfríos, lo mismo que lesiones óseas. La condición se resuelve al administrar antibióticos orales.
Escabiasis y otras erupciones pruriginosas La escabiasis, una erupción muy pruriginosa, tiene predilección por los glúteos, los genitales, el área periumbilical y los pliegues entre los dedos. Se ha descrito escabasis noruega, una forma psoriasiforme de costras gruesas, en pacientes con síndrome de Down y otros enfermos inmunosuprimidos, y en la actualidad se ve cada vez más en pacientes con SIDA. Las escamas de la escabiasis noruega contienen miles de ácaros que se ven con facilidad con el microscopio. Los surcos forman lesiones lineales de hasta 1 cm de largo. El ácaro causal, Sarcoptes scabiei, puede identificarse con el examen microscópico de los frotis de los surcos.
La foliculitis pustular eosinofílica y la erupción papular del SIDA son erupciones pruriginosas que afectan a los pacientes con infección por VIH. Ambas condiciones se caracterizan por prurito severo, pápulas del color de la piel y excoriaciones. Los pacientes con foliculitis pustular eosinofílica pueden desarrollar pústulas y pápulas eritematosas. Ambas condiciones responden al tratamiento con luz ultravioleta B.
Sarcoma de Kaposi
El sarcoma de Kaposi, una neoplasia vascular lentamente progresiva, se describió originalmente en varones ancianos italianos y judíos . Después se describió una forma más rápidamente progresiva en pacientes inmunosuprimidos con linfomas y pacientes con trasplante renal que recibían fármacos inmunosupresores. Se ha descrito una forma agresiva en pacientes con SIDA. El sarcoma de Kaposi clásico afecta típicamente las extremidades inferiores y progresa en forma gradual a otros sitios. Por el contrario, el sarcoma de Kaposi relacionado al SIDA puede ocurrir en cualquier superficie del organismo, incluyendo las mucosas. Se ha implicado al herpesvirus humano tipo 8 tanto en el sarcoma de Kaposi clásico como en el relacionado con el SIDA.22 Los tratamiento incluyen radioterapia, crioterapia e inyección intralesional con vinblastina.
Leucoplasia peluda oral
La leucoplasia peluda oral, otra condición que se ha descrito en pacientes infectados por VIH, consiste en pápulas blancas lineales sobre las superficies laterales de la lengua que causan la llamada apariencia peluda. La leucoplasia peluda oral puede distinguirse del algodoncillo oral porque las lesiones no pueden quitarse frotando, como en el algodoncillo.
OTROS ESTADOS DE INMUNODEFICIENCIA
Otros estados de inmunodeficiencia heredada o adquirida comparten diversas manifestaciones clínicas. La propensión a infecciones por monilia o infecciones bacterianas aumenta en trastornos como la enfermedad granulomatosa crónica y la neutropenia inducida por quimioterapia. También ocurren úlceras orales en la neutropenia cíclica y en la inmunosupresión inducida por quimioterapia.
Algunos medicamentos inmunosupresores tienen efectos cutáneos característicos. Los esteroides, cuando se usan por tiempo prolongado, causan fragilidad vascular, provocando púrpura esteroidea. También pueden provocar atrofia cutánea, formación de estrías y erupciones acneiformes. La ciclosporina se asocia con hipertricosis. La estomatitis aftosa es un efecto característico de numerosos medicamentos inmunosupresores, en especial de los agentes que disminuyen la función de la médula ósea.
Enfermedades infecciosas
Las manifestaciones cutáneas pueden ser importantes en diversas infecciones sistémicas, por ejemplo, los pacientes con septicemia pueden desarrollar coagulación intravascular diseminada (CID), que provoca hemorragias en la piel e infartos cutáneos. A continuación se describen características cutáneas clave de algunas infecciones sistémicas.
ENDOCARDITIS INFECCIOSA
Las manifestaciones cutáneas de la endocarditis infecciosa incluyen petequias, hemorragias en astilla (líneas rojas bajo la uña), nódulos de Osler (nódulos dolorosos y purpúricos en los cojinetes de los dedos de manos y pies) y lesiones de Janeway (máculas purpúricas no dolorosas de las palmas y plantas). Las lesiones cutáneas son causadas por embolias sépticas o vasculitis. El tratamiento de la infección subyacente causa resolución de las manifestaciones cutáneas.
SINDROME DE CHOQUE TOXICO
El síndrome de choque tóxico estafilocócico es una entidad descrita en forma reciente y que se detectó primero en mujeres menstruando que usaban tampones superabsorbentes. Es causado por una exotoxina producida por ciertas cepas de S. aureus.23 Se han implicado como causa las infecciones estafilocócicas en el hueso, tejidos blandos y otros sitios. Los pacientes desarrollan eritema difuso semejante al causado por quemadura solar con edema de las manos y pies, seguido de descamación de las palmas y plantas. También ocurre eritema de las mucosas, fiebre e hipotensión. Pueden desarrollarse síntomas gastrointestinales, menor función renal, elevación de las enzimas hepáticas, trombocitopenia y miositis.
SINDROME DE PIEL ESCALDADA POR ESTAFILOCOCO
El síndrome de piel escaldada por estafilococo (SSSS, por sus siglas en inglés, n. del t.) es causado por una toxina exfoliativa circulante producida por S. aureus fago grupo 11. La formación generalizada de bulas con grandes áreas de descamación es característica del padecimiento. Junto con el dolor, el eritema y la exfoliación de la piel, los pacientes tienen fiebre. No siempre es aparente el origen de la infección estafilocócica, y en ocasiones ésta se origina en una herida o un absceso oculto. Debido a que la infección estafilocócica suele estar lejos de la piel afectada, no crece S. aureus en el cultivo de piel.
El SSSS debe diferenciarse de la necrolisis epidérmica tóxica. Esta última afecta con frecuencia adultos y compromete las mucosas. El SSS suele afectar niños y respeta las mucosas. Además, la necrolisis epidérmica tóxica puede durar varias semanas y se asocia con una alta tasa de mortalidad, mientras que el SSSS dura pocos días y suele tener una buena evolución. Desde el punto de vista histológico, el SSSS muestra formación de bulas en la porción superior de la epidermis y la cavidad de la bula tiene células acantolíticas de apariencia normal y que flotan libres. En la necrolisis epidérmica tóxica ocurre formación de bulas en la capa basal de la epidermis y las células epidérmicas son necróticas.
FASCITIS NECROSANTE
La fascitis necrosante es causada por una infección anaeróbica mixta de una úlcera o una herida quirúrgica o traumática. La piel afectada está eritematosa, caliente y dolorosa, y desarrolla bulas hemorrágicas que se rompen para formar áreas de gangrena que crecen con rapidez y se extienden hacia abajo hasta la fascia. La debridación quirúrgica es indispensable para esta infección grave.24
MENINGOCOCCEMIA
La meningococcemia aguda puede ocurrir en epidemias o en casos aislados. Se desarrolla fiebre, cefalea y una erupción hemorrágica. Si no se trata los pacientes desarrollan CID con hemorragia importante, hipotensión y al final la muerte. El organismo causal, Neisseria meningititis, suele identificarse en el líquido cefalorraquídeo, pero también puede identificarse por frotis o cultivos de las lesiones de la piel o por hemocultivos.
FIEBRE ESCARLATINA
La fiebre escarlatina comienza con una faringitis causada por el estreptococo del grupo A. Se desarrolla una erupción generalizada 1 a 2 días después del inicio de la faringitis. La erupción se caracteriza por pápulas eritematosas puntiformes que es más fácil palpar que ver. Otras lesiones características incluyen una lengua en fresa y máculas petequiales lineales que ocurren en los pliegues del cuerpo (líneas de Pastia). Al disminuir la erupción aparece descamación de las palmas y las plantas. El tratamiento con penicilina causa resolución rápida de todos los síntomas.
INFECCION POR VIBRIO
La infección por Vibrio vulnificus se origina por traumatismos leves sufridos al nadar en lagos o el oceano o al limpiar alimentos del mar. Ocurre celulitis con linfangitis y bacteremia. En los pacientes con cirrosis hepática puede ocurrir infección después de comer ostras crudas. Estos pacientes desarrollan bulas hemorrágicas con leucopenia y coagulación intravascular diseminada.25
ENFERMEDAD DE LYME
La enfermedad de Lyme es causada por la espiroqueta Borrelia burgdorferi y se trasmite principalmente por la garrapata Ixodes dammini. La lesión cutánea característica, el eritema crónico migratorio, comienza como una mácula o una pápula eritematosa en el sitio de la mordedura de la garrapata. En días a semanas la lesión eritematosa se expande para formar un anillo rojo, con frecuencia con centro claro. Si no se tratan, las lesiones duran semanas o meses. Ocurre diseminación hematógena de las espiroquetas después de varias semanas, causando múltiples parches anulares de eritema crónico migratorio [ver figura 9]. Las complicaciones sistémicas incluyen una artritis aguda que afecta una o algunas articulaciones grandes pocas semanas después del inicio de los síntomas. Se desarrolla artritis erosiva crónica en alrededor del 10 por ciento de los pacientes. Pueden ocurrir síntomas neurológicos, incluyendo parálisis de Bell, lo mismo que complicaciones cardiacas, como insuficiencia cardiaca y alteraciones de la conducción.

|
| Figura 9 |
| Eritema crónico migratorio |
La enfermedad de Lyme puede prevenirse eliminando las garrapatas en las primeras 18 horas después de que se fijan. Una vez que se desarrollan los síntomas los antibióticos orales son eficaces para destruir a B. burgdorferi. Una vacuna que contiene una proteína elaborada por ingeniería genética a partir de la superficie de la bacteria evita la infección en la mayoría de las personas vacunadas.26
FIEBRE MANCHADA DE LAS MONTAÑAS ROCOSAS
La fiebre manchada de las Montañas Rocosas (FMMR) es una enfermedad trasmitida por garrapatas causada por Rickettsia rickettsii. Se caracteriza por inicio súbito de fiebre, calosfríos y cefalea. Alrededor de 4 días después se desarrolla una erupción eritematosa característica que se vuelve purpúrica en las muñecas y tobillos. La erupción se disemina hacia el centro para afectar extremidades, tronco y cara.
Debido a que la mortalidad de la FMMR es alta, ante la sospecha clínica la enfermedad debe tratarse de inmediato con cloranfenicol intravenoso o tetraciclinas. El diagnóstico puede establecerse después por biopsia de piel: la inmunofluorescencia con anticuerpos contra R. rickettsii muestra el organismo en las paredes de los vasos sanguíneos cutáneos. Las pruebas serológicas, como la reacción de Weil-Felix, pueden confirmar el diagnóstico después de la fase aguda de la enfermedad.
Enfermedades neurológicas
SINDROME DE NEVOS DE CELULAS BASALES
El síndrome de nevos de células basales es un trastorno autosómico dominante en el que los pacientes desarrollan carcinomas de células basales en una etapa temprana. Con el síndrome se asocian múltiples alteraciones esqueléticas y los individuos afectados pueden desarrollar quistes en la mandíbula. Ocurre calcificación en lámina de la hoz del cerebro, así como otras alteraciones neurológicas, incluyendo meduloblastomas.
SINDROME DE NEVO EPIDERMICO
El síndrome de nevo epidérmico se caracteriza por manifestaciones sistémicas como crisis convulsivas, retraso mental, ceguera y alteraciones esqueléticas asociadas con nevos epidérmicos grandes. Los nevos consisten en tiras pigmentadas largas que son lineales o en remolino y que afectan grandes áreas del cuerpo [ver figura 10].
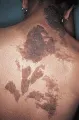
|
| Figura 10 |
| Síndrome del nevo epidérmico |
INCONTINENCIA DE PIGMENTO
La incontinencia de pigmento es un síndrome hereditario que afecta la piel y el sistema nervioso. El patrón de herencia es dominante ligado al X y es letal en fetos varones. La primera manifestación en la piel comienza a las semanas de nacimiento, en ocasiones in utero, y consiste en patrones lineales de lesiones vesiculobulosas. En semanas estas lesiones evolucionan a pápulas verrucosas y, eventualmente, a remolinos pigmentados. Aparte de los síntomas neurológicos, pueden ocurrir alteraciones oculares, alopecia cicatricial y malformaciones esqueléticas.
Hipomelanosis de Ito
La hipomelanosis de Ito, también llamada incontinencia de pigmento acromial, consiste en remolinos de hipopigmentación que se asocian con síntomas neurológicos en el 50 por ciento de los pacientes. Las lesiones cutáneas existen desde el nacimiento o se desarrollan en la infancia temprana. Además de las crisis convulsivas y el retraso mental, ocurren alteraciones esqueléticas y oculares.
NEUROFIBROMATOSIS
La neurofibromatosis es un padecimiento autosómico dominante común que afecta la piel y el sistema nervioso. Las lesiones cutáneas incluyen neurofibromas cutáneos, que son nódulos suaves, del color de la piel y con frecuencia pedunculados [ver figura 11]. Las manchas café con leche son planas, poco pigmentadas y hasta de varios centímetros de diámetro. Se encuentran seis o más manchas café con leche mayores de 1.5 cm de diámetro en la mayoría de los pacientes con neurofibromatosis tipo I (también llamada enfermedad de von Recklinhausen). Los neuromas plexiformes son tumores más grandes y profundos, que se asocian con hipertrofia ósea y de tejidos blandos. En una pequeña proporción de los tumores se originan neurofibrosarcomas. En la biopsia de piel las manchas café con leche contienen macromelanosomas, gránulos gigantes de pigmento en los melanocitos y los queratinocitos. También aparecen pecas axilares e inguinales como máculas pigmentadas que recuerdan manchas café con leche pequeñas en los sitios intertriginosos. Pueden encontrarse nódulos de Lisch (hamartomas pigmentados del iris) en la mayoría de los pacientes con neurofibromatosis.
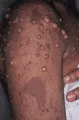
|
| Figura 11 |
| Neurofibromatosis tipo 1 |
Existen diferentes variantes de neurofibromatosis, incluyendo la neurofibromatosis segmentaria, en la que los pacientes desarrollan una distribución segmentaria de las manchas café con leche y neurofibromas cutáneos, y la neurofibromatosis tipo 2, que consiste en neuromas acústicos bilaterales sin nódulos de Lisch y con menos manchas café con leche que en el tipo 1. Los pacientes con neurofibromatosis tipo 2 pueden tener también algunos neurofibromas cutáneos. La neurofibromatosis tipos 1 y 2 son causadas por diferentes defectos genéticos, que se encuentran en los cromosomas 17 y 22, respectivamente.27,28
SINDROME DE SNEDDON
El síndrome de Sneddon es una enfermedad de la piel y del sistema nervioso causado por oclusión de arterias de tamaño pequeño a mediano en personas menores de 45 años. Las lesiones de la piel recuerdan al livedo reticularis y se han denominado livedo racemosa. Son frecuentes los episodios de isquemia transitoria o los eventos cerebrovasculares. El diagnóstico definitivo se realiza demostrando los cambios vasculares característicos en la biopsia de piel de los pacientes con datos neurológicos asociados.
ESCLEROSIS TUBEROSA
La esclerosis tuberosa es una enfermedad autosómico dominante que afecta la piel y el sistema nervioso central. Los pacientes afectados pueden desarrollar crisis convulsivas, retraso mental y lesiones cerebrales que pueden observarse en la TC. El adenoma sebáceo, la manifestación cutánea más característica de la esclerosis tuberosa, consiste en pápulas del color de la piel en la cara [ver figura 12a]. Otras lesiones son máculas hipopigmentadas denominadas manchas en hoja de fresno [ver figura 12b], lesiones hipopigmentadas más pequeñas conocidas como manchas en confeti, fibromas periungueales y subungueales (nódulos del color de la piel que se originan alrededor de las uñas de dedos de manos y pies) [ver figura 12c] y parches del color de la piel formados por tejido conectivo dérmico grueso. Se han identificado en los cromosomas 9 y 16 dos genes independientes que determinan la enfermedad.29
|
|
|
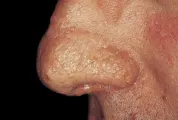

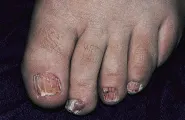
ENFERMEDAD DE FABRY
La enfermedad de Fabry es causada por una alteración de la alfa-galactosidasa A, causando depósito de glucoesfingolípidos en los tejidos corporales. El trastorno se hereda con un rasgo recesivo ligado al X. Los varones afectados suelen quejarse de dolor severo en las extremidades, con ardor en las palmas y plantas. Los episodios de dolor son temporales, pero los pacientes refieren parestesias persistentes en manos y pies.
Las lesiones cutáneas consisten en angioqueratomas, que son pápulas rojas o purpúricas puntiformes que recuerdan hemangiomas de cereza [ver figura 13]. Los angioqueratomas se encuentran con más frecuencia en el área periumbilical, pero pueden ocurrir también en las palmas, plantas, tronco, extremidades y mucosas. En los adultos se depositan glucoesfingolípidos en los vasos sanguíneos y en los órganos, afectando el corazón, válvulas cardiacas, arterias coronarias y riñones.

|
| Figura 13 |
| Enfermedad de Fabry |
POLIARTERITIS NODOSA
La poliarteritis nodosa es un trastorno inflamatorio que afecta las arterias musculares. Se forman aneurismas en muchas arterias, incluyendo las renales y del tejido subcutáneo. El diagnóstico de la forma sistémica de la poliarteritis puede hacerse demostrando aneurismas de las arterias renales en la angiografía renal.
La forma cutánea de poliarteritis nodosa se presenta principalmente como nódulos dolorosos en las extremidades inferiores.30 En los casos leves los pacientes pueden tener solo livedo reticularis, pero en los casos severos las lesiones cutáneas pueden ulcerarse. Puede existir polineuropatía asociada. Los pacientes con poliarteritis nodosa y microaneurismas tienen mayor incidencia de antigenemia de hepatitis B. A diferencia de otras vasculitis, suelen no tener anticuerpos anticitoplasma de neutrófilos.31
TRASTORNOS PERFORANTES
El término trastornos perforantes se refiere a varias condiciones que se caracterizan por salida de material dérmico a través de la epidermis. Estas lesiones se desarrollan con frecuencia asociadas con falla renal y diabetes mellitus.32 Las lesiones cutáneas se caracterizan por pápulas hiperqueratósicas con cráteres blancos centrales que histológicamente contienen material de la dermis. La colagenosis perforante reactiva, la foliculitis perforante y la enfermedad de Kyrle son todos ejemplos de trastornos perforantes asociados con insuficiencia renal.
CALCIFILAXIA
La calcifilaxia es un trastorno de pacientes con insuficiencia renal en la que áreas localizadas de la piel se vuelven necróticas por calcificación vascular. La calcifilaxia comienza con parches purpúricos dolorosos que pueden ser reticulados, recordando al livedo reticularis. Estos parches progresan a placas induradas que pueden ulcerarse, volviéndose necróticas [ver figura 14]. La calcifilaxia suele terminar en amputación o la muerte. La paratiroidectomía puede causar cicatrización de la piel afectada sin amputación.33
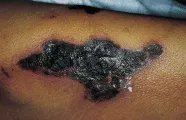
|
| Figura 14 |
| Calcifilaxia |
Enfermedades reumatológicas
DERMATOMIOSITIS
Las manifestaciones cutáneas más conocidas de la dermatomiositis, un trastorno inflamatorio del músculo y la piel, son las pápulas de Gottron y el eritema en heliotropo. Las pápulas de Gottron son máculas y pápulas descamativas eritematosas que ocurren en el dorso de los nudillos [ver figura 15]. El eritema en heliotropo consiste en eritema y edema periorbitario. Recientemente se han descrito lesiones en el cuero cabelludo, que pueden asociarse con alopecia.34 Las lesiones con frecuencia se confunden con dermatitis seborreica o psoriasis.
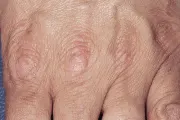
|
| Figura 15 |
| Dermatomiositis |
La asociación entre dermatomiositis y neoplasias sigue siento motivo de controversia. Se reportan neoplasias en el 15 a 50 por ciento de los pacientes, que suelen detectarse en la historia clínica y examen físico o en las pruebas de rutina, como mamografía, radiografía de tórax, examen de orina y examen de excremento para sangre oculta en heces. Rara vez se justifica la realización de más estudios, a menos que se encuentre evidencia de neoplasia en la evaluación inicial.35
Las clasificaciones de la dermatomiositis incluyen una variante juvenil caracterizada por calcificación de la piel o del músculo. La forma vasculítica en niños se complica por infartos cutáneos y ulceración, y por vasculitis gastrointestinal con dolor abdominal, hemorragia o perforación. Esta forma con vasculitis se asocia con mal pronóstico, y muchos de los pacientes fallecen por la enfermedad.
ESCLERODERMIA Y ENFERMEDADES SEMEJANTES
La esclerodermia incluye diversos síndromes diferentes que comparten una característica en común, la induración de la piel.
Esclerosis sistémica progresiva
La esclerosis sistémica progresiva, también conocida como esclerodermia sistémica, es una enfermedad frecuentemente fatal en la que los pacientes presentan fenómeno de Raynaud y esclerodactilia (induración de la piel de los dedos) [ver figura 16]. La induración cutánea puede volverse generalizada. La afección de la cara provoca una apariencia característica con fruncimiento de los labios y acartonamiento de la piel bajo la nariz que da la apariencia de pico de pájaro. Los pacientes con anticuerpos contra Scl-70 tienen mal pronóstico, falleciendo por enfermedad renal e hipertensión maligna. Los pacientes con anticuerpos anticentrómero tienen una variante de esclerodermia más lentamente progresiva conocida como síndrome de CREST, que se caracteriza por calcinosis cutánea, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia y telangiectasias. Con el tiempo se desarrolla fibrosis pulmonar, hipertensión pulmonar y falla cardiaca derecha.
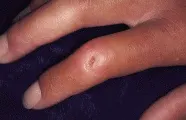
|
| Figura 16 |
| Esclerodactilia |
Morfea
La morfea, también denominada esclerodermia localizada, se caracteriza por parches bien delimitados de piel indurada que pueden volverse generalizados. Se distingue de la esclerosis sistémica progresiva por al ausencia de fenómeno de Raynaud, esclerodactilia o complicaciones sistémicas de la esclerodermia.
Existen innovaciones en el tratamiento tanto de la esclerosis sistémica progresiva como de la morfea. Se ha reportado que la exposición a psoralenos y luz ultravioleta de onda larga (PUVA) mejora la esclerosis sistémica progresiva y la morfea en forma dramática,36 y la exposición a UVA1 (el espectro de UVA más largo, de 340 a 400 nm) beneficia la esclerodermia localizada.37 Deben hacerse más estudios para confirmar la eficacia de estos tratamientos. Estudios anecdóticos han indicado que la minociclina puede ser eficaz en la esclerosis sistémica progresiva, pero se requieren estudios controlados al respecto.38
Enfermedad injerto contra huésped
Al volverse más común el trasplante de órganos ha aumentado en frecuencia otra enfermedad parecida a la esclerodermia, la enfermedad injerto contra huésped, en especial después del trasplante de médula ósea. Existen dos fases en la enfermedad injerto contra huésped. La primera, la fase aguda, se desarrolla 10 a 40 días después del trasplante y consiste en una erupción macular y papular eritematosa que suele asociarse con fiebre, hepatomegalia, linfadenopatía o síntomas gastrointestinales. La enfermedad injerto contra huésped crónica se desarrolla 3 meses después del trasplante y consiste en pápulas purpúricas que recuerdan liquen plano [ver figura 17]. Lo más característico son los cambios cutáneos esclerodermatosos con telangiectasias, hiperpigmentación reticulada y alopecia. Tanto la ciclosporina como la PUVA han demostrado ser útiles en la prevención y tratamiento de la enfermedad injerto contra huésped.39,40

|
| Figura 17 |
| Reacción injerto contra huésped |
Fascitis eosinofílica
Ocurre endurecimiento de la piel parecido a la esclerodermia en la fascitis eosinofílica. Típicamente se desarrollan pliegues de la piel de las extremidades asociados con dolor. A diferencia de la esclerosis sistémica progresiva, no ocurre fenómeno de Raynaud. El diagnóstico definitivo requiere biopsia de la piel y la fascia suprayacente al músculo afectado. En algunos casos de fascitis eosinofílica se desarrollan alteraciones hematológicas, incluyendo anemia aplástica, trombocitopenia, enfermedad de Hodgkin y leucemias.41
Síndrome de mialgia eosinofilia
Algunos pacientes con diagnóstico erróneo de fascitis eosinofílica habían ingerido L-triptofano, que se ha asociado con el síndrome de mialgia eosinofilia. Como resultado ocurre dolor y debilidad musculares severas, junto con fiebre, eosinofilia y diversos síntomas sistémicos. Las manifestaciones cutáneas más comunes son eritema y edema.42
LUPUS ERITEMATOSO GENERALIZADO
Existen muchas manifestaciones cutáneas en el lupus, incluyendo fenómeno de Raynaud, el llamado lupus discoide (caracterizado por lesiones cicatriciales redondas con hipopigmentación central y un borde de hiperpigmentación), eritema malar, fotosensibilidad, alopecia y úlceras mucosas. Al aprender más sobre lupus el espectro de las enfermedades cutáneas asociadas con este trastorno continúa expandiéndose. El lupus cutáneo subagudo, una variante caracterizada serológicamente por anticuerpos anti-Ro y anti-La, se asocia con lesiones psoriasiformes anulares de la piel [ver figura 18].
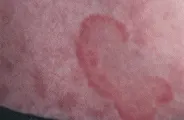
|
| Figura 18 |
| Lupus eritematoso cutáneo subagudo |
Síndrome de anticuerpos anticardiolipina
El síndrome de anticuerpos anticardiolipina, que puede ocurrir en los pacientes con lupus, se ha descrito en pacientes que sufren episodios repetidos de flebitis, trombosis arteriales y abortos de repetición. Son comunes los infartos cutáneos. Los pacientes tienen serología falsa positiva para sífilis y tienen anticoagulante lúpico circulante. Los anticuerpos antifosfolípido circulantes son la característica serológica de este síndrome recién descrito.43
Livedo vasculitis
La livedo vasculitis, otra entidad que se ha asociado con el lupus, se caracteriza por úlceras dolorosas y recurrentes en las piernas y tobillos. Las úlceras sanan dejando cicatrices escleróticas blancas. Los pacientes afectados con frecuencia tienen livedo reticularis. Esta condición, también conocida como atrofia blanca, se ha atribuido a procesos trombóticos más que a depósito de complejos inmunes o vasculitis leucocitoclástica.44
Lupus neonatal
El lupus neonatal es un síndrome caracterizado por máculas y pápulas eritematosas anulares que ocurren en la cara de niños recién nadidos. El trastorno se ha atribuido al paso transplacentario de anticuerpos anti-Ro y en ocasiones anti-La. Las madres con frecuencia están asintomáticas, pero pueden tener lupus o síndrome de Sjögren. El bloqueo cardiaco congénito es la complicación más seria de este padecimiento.
Bibliografía
- Mana J, Marcoval J, Graells J, et al: Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol 133:882, 1997
- McNiff JM, Cooper D, Howe G, et al: Lymphomatoid granulomatosis of the skin and lung: an angiocentric T-cell-rich B-cell lymphoproliferative disorder. Arch Dermatol 132: 1464, 1996
- Davis MD, Daoud MS, McEvoy MT, et al: Cutaneous manifestations of Churg-Strauss syndrome: a clinicopathologic correlation. J Am Acad Dermatol 37:199, 1997
- Leung DY, Sullivan KE, Brown-Whitehorn TF, et al: Association of toxic shock syndrome toxin-secreting and exfoliative toxin-secreting Staphylococcus aureus with Kawasaki syndrome complicated by coronary artery disease. Pediatr Res 42:268, 1997
- Lebwohl MG, Distefano D, Prioleau PG, et al: Pseudoxanthoma elasticum and mitral valve prolapse. N Engl J Med 307:228, 1982
- Lebwohl M, Halperin J, Phelps RG: Brief report: occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N Engl J Med 329:1237, 1993
- Lebwohl M, Phelps RG, Yannuzzi L, et al: Diagnosis of pseudoxanthoma elasticum by scar biopsy in patients without characteristic skin lesions. N Engl J Med 317:347, 1987
- Struk B, Neldner KH, Rao VS, et al: Mapping of both autosomal recessive and dominant variants of pseudoxanthoma elasticum to chromosome 16p13.1. Hum Mol Genet 6: 1823, 1997
- Bull RH, Fenton DA, Mortimer PS: Lymphatic function in the yellow nail syndrome. Br J Dermatol 134:307, 1996
- Verrotti A, Chiarelli F, Amerio P, et al: Necrobiosis lipoidica diabeticorum in children and adolescents: a clue for underlying renal and retinal disease. Pediatr Dermatol 12:220, 1995
- Rho YW, Suhr KB, Lee JH, et al: A clinical observation of scleredema adultorum and its relationship to diabetes. J Dermatol 25:103, 1998
- Hecker MS, Lebwohl MG: Recalcitrant pyoderma gangrenosum: treatment with thalidomide. J Am Acad Dermatol 38:490, 1998
- Barax C, Lebwohl M, Phelps RG: Multiple hamartoma syndrome. J Am Acad Dermatol 17:342, 1987
- Collin P, Pukkala E, Reunala T: Malignancy and survival in dermatitis herpetiformis: a comparison with coeliac disease. Gut 38:528, 1996
- Nakagawa H, Koyama K, Tanaka T, et al: Localization of the gene responsible for Peutz-Jeghers syndrome within a 6-cM region of chromosome 19p13.3. Hum Genet 102:203, 1998
- Christiano AM, LaForgia S, Paller AS, et al: Prenatal diagnosis for recessive dystrophic epidermolysis bullosa in 10 families by mutation and haplotype analysis in the type VII collagen gene (COL7A1). Mol Med 2:59, 1996
- Topar G, Staudacher C, Geisen F, et al: Urticaria pigmentosa: a clinical, hematopathologic, and serologic study of 30 adults. Am J Clin Pathol 109:279, 1998
- Horny HP, Ruck P, Krober S, et al: Systemic mast cell disease (mastocytosis): general aspects and histopathological diagnosis. Histol Histopathol 12:1081, 1997
- Pottage JC Jr, Kessler HA: Herpes simplex virus resistance to acyclovir: clinical relevance. Infect Agents Dis 4:115, 1995
- Lalezari J, Schacker T, Feinberg J, et al: A randomized, double-blind, placebo-controlled trial of cidofovir gel for the treatment of acyclovir-unresponsive mucocutaneous herpes simplex virus infection in patients with AIDS. J Infect Dis 176:892, 1997
- Koehler JE, Quinn FD, Berger TG, et al: Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis. N Engl J Med 327:1625, 1992
- Kennedy MM, Cooper K, Howells DD, et al: Identification of HHV8 in early Kaposi's sarcoma: implications for Kaposi's sarcoma pathogenesis. Mol Pathol 51:14, 1998
- Parsonnet J: Nonmenstrual toxic shock syndrome: new insights into diagnosis, pathogenesis, and treatment. Curr Clin Top Infect Dis 16:1, 1996
- Green RJ, Dafoe DC, Raffin TA: Necrotizing fasciitis. Chest 110:219, 1996
- Hally RJ, Rubin RA, Fraimow HS, et al: Fatal Vibrio parahemolyticus septicemia in a patient with cirrhosis: a case report and review of the literature. Dig Dis Sci 40:1257, 1995
- Steere AC, Sikand VK, Meurice F, et al: Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvant: Lyme Disease Vaccine Study Group. N Engl J Med 339:209, 1998
- Upadhyaya M, Osborn MJ, Maynard J, et al: Mutational and functional analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet 99:88, 1997
- De Klein A, Riegman PH, Bijlsma EK, et al: A G-A transition creates a branch point sequence and activation of a cryptic exon, resulting in the hereditary disorder neurofibromatosis 2. Hum Mol Genet 7:393, 1998
- Soucek T, Holzl G, Bernaschek G, et al: A role of the tuberous sclerosis gene-2 product during neuronal differentiation. Oncogene 16:2197, 1998
- Daoud MS, Hutton KP, Gibson LE: Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol 136:706, 1997
- Guillevin L, Lhote F, Amouroux J, et al: Antineutrophil cytoplasmic antibodies, abnormal angiograms and pathological findings in polyarteritis nodosa and Churg-Strauss syndrome: indications for the classification of vasculitides of the polyarteritis nodosa group. Br J Rheumatol 35:958, 1996
- Poliak S, Lebwohl MG, Parris A, et al: Reactive perforating collagenosis associated with diabetes mellitus. N Engl J Med 306:81, 1982
- Angelis M, Wong LL, Myers SA, et al: Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery 122:1083 (discussion: 1089), 1997
- Kasteler JS, Callen JP: Scalp involvement in dermatomyositis: often overlooked or misdiagnosed. JAMA 272:1939, 1994
- Drake LA, Dinehart SM, Farmer ER, et al: Guidelines of care for dermatomyositis. American Academy of Dermatology. J Am Acad Dermatol 34:824, 1996
- Kanekura T, Fukumaru S, Matsushita S, et al: Successful treatment of scleroderma with PUVA therapy. J Dermatol 23:455, 1996
- Kerscher M, Volkenandt M, Gruss C, et al: Low-dose UVA phototherapy for treatment of localized scleroderma. J Am Acad Dermatol 38:21, 1998
- Le CH, Morales A, Trentham DE: Minocycline in early diffuse scleroderma. Lancet 352:1755, 1998
- Zikos P, van Lint MT, Frassoni F, et al: Low transplant mortality in allogeneic bone marrow transplantation for acute myeloid leukemia: a randomized study of low-dose cyclosporin versus low-dose cyclosporin and low-dose methotrexate. Blood 91:3503, 1998
- Vogelsang GB, Wolff D, Altomonte V, et al: Treatment of chronic graft-versus-host disease with ultraviolet irradiation and psoralen (PUVA). Bone Marrow Transplant 17:1061, 1996
- Kim SW, Rice L, Champlin R, et al: Aplastic anemia in eosinophilic fasciitis: responses to immunosuppression and marrow transplantation. Haematologia (Budap) 28:131, 1997
- Gordon M, Lebwohl M, Phelps RG, et al: Eosinophilic fasciitis associated with tryptophan ingestion: a manifestation of eosinophilia-myalgia syndrome. Arch Dermatol 127:217, 1991
- Derksen RH: Clinical manifestations and management of the antiphospholipid syndrome. Lupus 5:167, 1996
- McCalmont CS, McCalmont TH, Jorizzo JL, et al: Livedo vasculitis: vasculitis or thrombotic vasculopathy? Clin Exp Dermatol 17:4, 1992
- Brucato A, Franceschini F, Buyon JP: Neonatal lupus: long-term outcomes of mothers and children and recurrence rate. Clin Exp Rheuatol 15:467, 1997