Inmunología
⭳ Abrir artículo (PDF)590.5 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
X ENFERMEDAD POR COMPLEJOS INMUNES
- Fisiología o fisiopatología
- FORMACION DE UNA RED ANTIGENO-ANTICUERPO
- DEPURACION FISIOLOGICA DE LOS COMPLEJOS INMUNES
- DEPOSITO ABERRANTE DE LOS COMPLEJOS INMUNES
- LESION TISULAR MEDIADA POR COMPLEJOS INMUNES: REACCION DE ARTHUS
- OTROS EFECTOS DE LOS COMPLEJOS INMUNES
- Enfermedad por complejos inmunes localizada
- Enfermedad sistémica por complejos inmunes
- ENFERMEDAD DEL SUERO
- NEFRITIS CRONICA POR COMPLEJOS INMUNES
- OTRAS ENFERMEDADES POR COMPLEJOS INMUNES
- Detección de complejos inmunes circulantes
- Tratamiento
X ENFERMEDAD POR COMPLEJOS INMUNES
DR. WILLY F. PIESSENS
El término enfermedad por complejos inmunes se refiere a un grupo de padecimientos cuya patogenia incluye el daño tisular por reacciones de complejos inmunes. Debido a que siempre que los anticuerpos interactúan con antígenos exógenos o endógenos se forman complejos inmunes potencialmente dañinos, las enfermedades por complejos inmunes son heterogéneas, con causas y manifestaciones clínicas muy diversas.
Fisiología o fisiopatología
FORMACION DE UNA RED ANTIGENO-ANTICUERPO
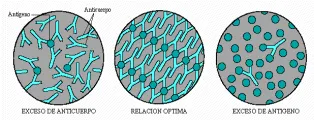
Varios factores determinan el tamaño y solubilidad de un complejo inmune formado in vitro, incluyendo la relación molar de anticuerpo a antígeno, las propiedades intrínsecas de estas dos moléculas y el medio en que la reacción tiene lugar. Los estudios clásicos sobre reacciones de inmunoprecipitación ilustran de la mejor manera la contribución de las cantidades relativas de antígeno y anticuerpo al tamaño del complejo. Los complejos más grandes, que son los menos solubles, se forman cuando la relación molar de anticuerpo a antígeno se aproxima a 1 [ver figura 1]. Esta región se conoce como zona de equivalencia, en la que no puede detectarse ni antígeno ni anticuerpo libre en el sobrenadante de la reacción de precipitación. Los complejos que se forman cuando existe exceso de antígeno o de anticuerpo son más pequeños que los formados en la zona de equivalencia y causan precipitados de menor tamaño [ver figura 2].
La valencia antigénica, o número de epítopes
antigénicos en cada molécula, determina el número de
sitios posibles en donde el antígeno y el anticuerpo pueden interactuar
para formar una red de tres dimensiones. Debido a que la mayoría de los
antígenos son multivalentes y cada molécula de anticuerpo tiene
dos sitios de combinación, es posible construir una red en la que las
moléculas de antígeno y anticuerpo se alternan [ver figura
1]. El tamaño teórico de estos complejos es infinito, pero en
la práctica se limita por las cantidades de antígeno y
anticuerpo. La clase de la imunoglobulina y la afinidad del anticuerpo por el
antígeno también influyen en el tamaño de un complejo
inmune. Los complejos que contienen IgM son más grandes y se precipitan
con más rapidez que los que contienen anticuerpos de IgG o IgA. Los
anticuerpos con baja afinidad para un antígeno producen complejos
más pequeños que los anticuerpos con alta afinidad.
El complemento es otro determinante importante del tamaño de un complejo inmune.2 La interacción de un anticuerpo IgM o IgG fijador de complemento con un antígeno induce cambios conformacionales en la porción Fc de la molécula de inmunoglobulina que desencadenan la activación del complemento a través de la vía clásica. La unión de C1 a la inmunoglobulina, que inicia la reacción del complemento, retrasa las interacciones Fc-Fc y en circunstancias normales provoca la formación de complejos que son menores que los que se formarían en ausencia de complemento [ver figura 3]. Los complejos que contienen IgA, una clase de anticuerpos que no fijan C1, pueden activar la vía alterna del complemento, lo que ocasiona depósito de C3b. LA molécula C3b debilita las fuerzas que mantienen juntos al antígeno y al anticuerpo. El grado de fijación varía con el tamaño del complejo (los complejos más grandes fijan más complemento que los pequeños) y con el isotipo del anticuerpo (IgM>IgG>IgA). Por lo general los complejos que contienen IgE no fijan complemento.
DEPURACION FISIOLOGICA DE LOS COMPLEJOS INMUNES
En condiciones fisiológicas los complejos inmunes que se forman directamente dentro de los tejidos son transportados por los linfáticos para drenar en los ganglios linfáticos,3 mientras que los complejos de la sangre son eliminados de la circulación por el sistema reticuloendotelial (SRE). Los fagocitos mononucleares en los ganglios linfáticos, bazo e hígado fagocitan y degradan los complejos.
La opsonización de los complejos inmunes (la unión covalente de muchas moléculas C3b al complejo antígeno-anticuerpo cuando se activa complemento por la vía clásica) facilita la depuración de los complejos inmunes de la circulación. La opsonización permite que las células que poseen un receptor específico para C3b, el receptor 1 de complemento (CR1, por sus siglas en inglés, n. del t.), fijen a los complejos opsonizados (cubiertos por C3b) y los entreguen al SRE.2,4 El CR1 está presente en muchos tipos de células, pero más del 90 porciento de los CR1 en la sangre que encuentran en las membranas de los eritrocitos. Por lo tanto, los eritrocitos son los principales transportadores de complejos inmunes al SRE.5 Este modo de transporte parece proteger al endotelio vascular del daño mediado por complejos inmunes.
Varios factores facilitan la transferencia de complejos inmunes de los eritrocitos a los fagocitos del SRE en el bazo y el hígado. La activación de la vía alterna del complemento induce la liberación de los complejos unidos a los eritrocitos, la unión de los complejos inmunes a los eritrocitos se debilita cuando el factor I del complemento en el plasma separa al C3b unido al complejo. Los fagocitos poseen más receptors CR1 que los eritrocitos. Además, los fagocitos contienen receptores para la porción Fc de la IgG, que no existen en los eritrocitos [ver figura 4]. Los receptores Fc en las células del SRE no solo capturan los complejos inmunes unidos a los eritrocitos, sino también eliminan a los complejos inmunes que contienen IgG libres de la circulación.6 Los complejos grandes y opsonizados son eliminados en forma eficiente de la sangre en minutos. Los complejos pequeños son malos activadores del complemento y pueden continuar circulando durante muchos días.
DEPOSITO ABERRANTE DE LOS COMPLEJOS INMUNES
Las alteraciones en la depuración fisiológica de los complejos inmunes pueden causar depósitos aberrantes en los vasos sanguíneos y los tejidos circundantes. Varios procesos estimulan la liberación local de sustancias vasoactivas que aumentan la permeabilidad vascular. Los antígenos en los complejos circulantes pueden interactuar con anticuerpos IgE específicos y anticuerpos acoplados con receptores Fc de los basófilos circulantes , provocando la liberación de sustancias como histamina y factor activador de plaquetas (FAP). El FAP, las anafilotoxinas C3a y C5a generadas durante la activación del complemento o los complejos inmunes per se causan agregación y degranulación plaquetaria, con liberación de histamina y serotonina. Todos estos factores facilitan el depósito de complejos inmunes en las paredes de los vasos sanguíneos y migración de los complejos hacia los tejidos. Estas sustancias vasoactivas también provocan la urticaria que ocurre en la enfermedad por complejos inmunes.
Los depósitos pueden ocurrir en cualquier parte del organismo, pero las lesiones tienden a localizarse en áreas en donde las plaquetas se adhieren con más frecuencia al endotelio por la mayor presión sanguínea y la turbulencia. Varios factores adicionales pueden explicar porqué algunos órganos, como el riñón, se afectan con más frecuencia que otros.7 La combinación de flujo sanguíneo rápido, filtración intensa y presión sanguínea alta en los glomérulos renales facilita el depósito de complejos inmunes. Los receptores para los componentes activados del complemento en los podocitos de los glomérulos normales pueden causar que los complejos inmunes se concentren en este sitio.8 Estos receptores están también presentes en el plexo coroideo, otro sitio común de depósitos de complejos inmunes. Los receptores para los componentes activados del complemento en los podocitos se concentran en este sitio.8 Estos receptores están presentes también en el plexo coroideo, otro sitio común de depósito de complejos inmunes. Los complejos son atrapados con facilidad en la membrana basal glomerular porque el endotlio vascular renal es fenestrado (muy poroso) y el intersticio renal contiene receptores para Fc. Además, las membranas basales, se encuentren en el glomérulo renal o en la piel, tienen carga negativa, por lo que retienen los complejos inmunes con carga positiva por más tiempo que los neutros o con carga negativa. El efecto de la carga puede explicar el depósito frecuente de complejos inmunes en las regiones subendoteliales del glomérulo y en la unión dermoepidérmica de la piel.
Cuando la capacidad de los eritrocitos para actuar como transportadores de los complejos inmunes está alterada, los complejos se depositan con más facilidad en los vasos sanguíneos y no son eliminados por los fagocitos mononucleares del bazo y el hígado. Esta alteración ocurre en los pacientes que tienen lupus eritematoso generalizado (LEG), un padecimiento en el que los eritrocitos tienen menor número de receptores C3b.9 De igual modo, cuando los fagocitos en el hígado y el bazo están disminuidos en número, bloqueados por haber captado antes una partícula, o deficientes en complemento o receptores Fc, se depositan complejos dañinos en otros órganos. La deficiencia del receptor C3b se ha asociado con vasculitis inmune y paniculitis en la piel de pacientes con LEG.10
La actividad funcional de los receptores Fc sobre las células del SRE puede evaluarse por medio de eritrocitos autólogos radiomarcados y cubiertos con anticuerpos IgG anti-eritrocito in vitro. Cuando se inyectan estás células al paciente, la velocidad de desaparición de las mismas correlaciona con la actividad del receptor Fc. Los estudios de pacientes con LEG muestran un defecto importante en la depuración específica del receptor Fc que correlaciona con la actividad de la enfermedad.11 La función Fc de los macrófagos también está alterada en pacientes con nefropatía terminal.12
LESION TISULAR MEDIADA POR COMPLEJOS INMUNES: REACCION DE ARTHUS
La reacción de Arthus es un proceso hemorrágico y necrótico agudo que ocurre en dos a cuatro horas después de que se inyecta el antígeno en la piel u otros tejidos de un animal inmunizado. Esta reacción se considera el prototipo de la lesión tisular aguda y localizada mediada por complejos inmunes, y se ha usado para estudiar la secuencia de eventos por los que el depósito de complejos inmunes provoca daño tisular. Al inicio se desarrolla una intensa infiltración de leucocitos polimorfonucleares dentro y alrededor de los vasos sanguíneos. La alteración de la pared vascular causa que los eritrocitos salgan hacia los tejidos. Posteriormente linfocitos, células plasmáticas y monocitos infiltran las lesiones y ocurre proliferación de las células endoteliales. Puede detectarse anticuerpo, antígeno y complemento en las lesiones por medio de microscopía inmunofluorescente [ver figura 5]. Los estudios de esta reacción indican que los componentes del complemento y los neutrófilos son esenciales para la formación de estas lesiones.
La reacción de Arthus comienza en el tejido cercano a un vaso
sanguíneo cuando los complejos inmunes precipitados fijan C1q, activando
así al complemento a través de la vía clásica
[ver figura 6]. Esta reacción genera la ruptura
proteolítica de productos como C3a, C3b y C5a. C3a y C5a desencadenan la
liberación por las células cebadas de diversos mediadores
solubles como histamina, sustancia de reacción lenta de la anafilaxia y
factor activador de plaquetas. Los mediadores liberados por las células
cebadas y las plaquetas actúan en conjunto para aumentar la
permeabilidad vascular, lo que permite la entrada de más anticuerpo y
complemento al sitio y amplifica la reacción. Algunos de estos
mediadores pueden causar también contracción del músculo
liso e hipotensión.
C3b, que se forma cuando los complejos inmunes activan complemento, se fija con rapidez a la membrana basal de los vasos. La fijación de los neutrófilos a las estructuras cubiertas por C3b a través del CR1 causa también liberación de gránulos lisosómicos en ausencia de fagocitosis. Por último, C3e, un fragmento derivado de la mayor degradación de C3b, estimula la neutrofilia reactiva al liberar células de la médula ósea.
C5a, así como citocinas como la interleucina-8 (IL-8) y el factor de necrosis tumoral-alfa (FNT-alfa), liberados por las células cebadas, son potentes quimiotácticos para los neutrófilos. Estas células se acumulan en el sitio de reacción, fijan complejos antígeno-anticuerpo a través de sus receptores Fc y fagocitan los complejos. Durante este proceso los neutrófilos liberan un amplio rango de productos que pueden dañar los componentes tisulares. Estos productos incluyen moléculas oxidantes y radicales como el H2O2 y los aniones superóxido, péptidos catiónicos que aumentan la permeabilidad capilar, causan contracción del músculo liso y desecadenan mayor liberación de los mediadores de células cebadas, y enzimas que degradan las membranas basales en los riñones y vasos sanguíneos, glucosaminoglicanos y cartílago en las articulaciones, fibras elásticas en los vasos sanguíneos y colágena en muchos tejidos. La liberación de estas hidrolasas puede ser inhibida en parte por agentes que aumentan el monofosfato cíclico de adenosina (AMPc), como la prostaglandina E1. La lesión tisular causada por el depósito de complejos inmunes también está mediada en parte por exceso de óxido nítrico liberado en forma local (NO), un mediador potente y pleiotrópico secretado por macrófagos, neutrófilos y células endoteliales activados. El FAP induce sintetasa de óxido nítrico, una enzima que genera NO.13
El modelo tradicional sostiene que la reacción de Arthus inicia cuando los complejos inmunes activan al complemento en los tejidos a través de la vía clásica. Se generan fragmentos quimiotácticos del complmento para atraer neutrófilos y estimular a éstas células y a las células cebadas a liberar diversos productos solubles que causan daño tisular. Un estudio reciente pone en duda la secuencia de los eventos. Los resultados del estudio sugieren que la reacción de Arthus puede iniciarse por la interacción de los complejos inmunes con receptores Fc de las células y ser después amplificada por productos activados del complemento y mediadores celulares.14 Otros investigadores destacan la contribución de las citocinas quimiotácticas y las moléculas de adhesión en la extravasación y activación de neutrófilos15 e indican que el antagonista del receptor de la interleucina-1 (IL-1) modula el daño tisular mediado por complejos inmunes.16 Estas observaciones han originado nuevos enfoques de tratamiento de los síndromes de complejos inmunes en modelos animales [ver adelante, Tratamiento].
OTROS EFECTOS DE LOS COMPLEJOS INMUNES
En condiciones fisiológicas los complejos antígeno-anticuerpo tienen funciones inmunorreguladoras importantes. El antígeno es transportado a través de la linfa hacia los centros germinales de los ganglios linfáticos regionales, en donde forma un complejo con anticuerpo y complemento. La retención del antígeno en los centros germinales es necesaria para el desarrollo de células B de memoria. Las interacciones idiotipo-antidiotipo, un tipo especializado de formación de complejo inmune, parecen ser reguladores generales de las respuestas inmunes. Los complejos inmunes pueden inducir también la activación o la tolerancia a inyecciones subsecuentes de antígeno solo, un hecho relevante para la programación de las vacunas.17
La interacción dependiente de Fc o de complemento de las células con blancos específicos puede ser inhibida por complejos inmunes que compiten por los mismos receptores. Esta inhibición, o bloqueo, parece particiar en el escape de las células tumorales y parásitos de la citotoxicidad mediada por células dependientes de anticuerpos. En el suero de los pacientes con fibrosis quística existen niveles altos de complejos inmunes que contienen IgG2 y que bloquean los receptores Fc y así inhiben la fagocitosis de Pseudomonas aeruginosa. Los complejos inmunes en el suero de estos pacientes pueden activar también células T para destruir células B que producen anticuerpos opsonizantes específicos. Estos dos mecanismos pueden ser responsables de la infección crónica por Pseudomonas que se presenta en las vías respiratorias en la fibrosis quística.18,19
Enfermedad por complejos inmunes localizada
Los antígenos que desencadenan la enfermedad por complejos inmunes localizada pueden ser endógenos estructurales, celulares, autoantígenos tisulares secretados, o antígenos exógenos que se concentran en ciertos órganos [ver tabla 1]. Los síntomas clínicos que ocasionan varían de acuerdo con la localización de la reacción por complejo inmune. El complemento puede tener un papel muy importante en la patogenia de algunas enfermedades por complejos inmunes localizadas, como lo ilustran los estudios experimentales sobre miastenia gravis. En este modelo los anticuerpos reaccionan con receptores de acetilcolina, pero la destrucción inflamatoria de las placas motoras terminales no ocurre cuando se evita la activación del complemento.
Por lo general no ocurre una reacción de Arthus cuando se inyecta a una persona un antígeno extraño porque no existe suficiente anticuerpo circulante contra el antígeno. Sin embargo, las concentraciones locales de antígeno en una persona sensibilizada que tiene anticuerpos específicos pueden causar lesión tisular mediada por complejos inmunes [ver tabla 1]. La inflamación que se desarrolla después de una inyección de refuerzo de toxoide tetánico puede ser en parte una reacción de tipo Arthus. Cuando las personas que tienen anticuerpos específicos contra diferentes antígenos micóticos o proteínas animales inhalan estos antígenos, las reacciones tipo Arthus en el pulmón causan neumonitis por hipersensibilidad. Los síntomas consisten en fiebre, tos y disnea, que comienzan cuatro a seis horas después de la exposición al antígeno específico, alcanzan su máximo después de 12 a 24 horas y suelen resolverse en dos a tres días. Las radiografías de los pulmones durante la fase aguda muestran edema intersticialy, en ocasiones, infiltrados en parches. La biopsia pulmonar revela infiltración por granulocitos y linfocitos. En la enfermedad pulmonar crónica por complejos inmunes se observan los cambios típicos del enfisema (infiltración con macrófagos y pérdida de las fibras de elastina y colágena). La enfermedad por complejos inmunes de los pulmones también ocurre en niños que han recibido vacunas por virus muertos que inducen formación de anticuerpos pero no producen inmunidad protectora, como la vacuna del sarampión con virus muertos. Cuando estos niños son expuestos en una etapa posterior a virus vivos los antígenos se combinan con los anticuerpos y producen lesión por complejos inmunes.
Enfermedad sistémica por complejos inmunes
ENFERMEDAD DEL SUERO
La enfermedad del suero fue descrita por primera vez a principios del siglo XX en pacientes que recibieron suero de caballo para el tratamiento de enfermedades infecciosas. La enfermedad del suero es común en pacientes que reciben inmunoglobulina antitimocítica equina como tratamiento para la falla de la médula ósea. También ocurre en pacientes infectados por el virus de inmunodeficiencia humana (VIH) que desarrollan anticuerpos contra suero fetal de ternera después de infusiones de linfocitos singénicos20 y en algunos pacientes tratados con estreptocinasa.21
Varios fármacos no proteináceos pueden provocar enfermedad del suero, incluyendo penicilina, oro, penicilamina, sulfonamidas, tiouracilos, hidantoínas, tiacidas, fenilbutazona, ácido aminosalicílico y estreptomicina. Estos medicamentos parecen actuar como haptenos que inducen respuestas de anticuerpos cuando se acoplan con proteínas transportadoras. La administración repetida de proteínas extrañas y fármacos de depósito o vida media muy larga, como penicilamina y oro, puede inducir enfermedad del suero crónica.
Típicamente, ocho a 12 días después de la administración de una gran cantidad de suero de especies extrañas el paciente muestra fiebre, malestar, ganglios linfáticos aumentados de tamaño y dolorosos, articulaciones dolorosas e inflamadas, erupciones cutáneas, molestias gastrointestinales y crecimiento del bazo. Los estudios de laboratorio pueden mostrar leucocitosis, proteinuria, menor actividad del complemento sérico y, en ocasiones, eosinofilia. Los síntomas suelen desaparecer después de una semana, momento en el que pueden detectarse anticuerpos contra los antígenos extraños en el suero. El inicio de los síntomas ocurre en forma más temprana después de una segunda inyección de suero. la enfermedad del suero suele ser benigna y autolimitada, sus principales complicaciones son vasculitis, glomerulonefritis, artritis y neuritis. En ocasiones ocurre síndrome de Guillain-Barré. Aunque los signos y síntomas neurológicos pueden durar un tiempo prolongado, la recuperación suele ser completa.
La patogenia de la enfermedad del suero en humanos es semejante a la descrita en modelos animales [ver figura 7].22 La enfermedad clínica coincide con el desarrollo de niveles muy altos de complejos inmunes circulantes y disminuye en forma importante los niveles séricos de C3, C4 y actividad del complemento hemolítico. Pueden detectarse inmunoglobulinas y C3 por microscopía con inmunofluorescencia en las paredes de los vasos cutáneos pequeños en la mayoría de los pacientes con enfermedad del suero aguda. Una banda serpiginosa no habitual de eritema en las palmas y plantas puede ser el primer signo de esta entidad.
NEFRITIS CRONICA POR COMPLEJOS INMUNES
Debido a que los glomérulos renales son especialmente vulnerables a la lesión inmune mediada por complejos, la glomerulonefritis es una característica frecuente de muchas enfermedades por complejos inmunes y con frecuencia ocurre en ausencia de otras manifestaciones clínicas. Los complejos formados en la circulación suelen depositarse en las áreas mesangiales y endoteliales y producir un patrón irregular típico en la inmunofluorescencia ('lumpy-bumpy').
OTRAS ENFERMEDADES POR COMPLEJOS INMUNES
Infecciones
La glomerulonefritis y otras manifestaciones de vasculitis pueden ocurrir en asociación con muchas infecciones bacterianas persistentes (v.gr., endocarditis bacteriana subaguda), infecciones de derivaciones arteriovenosas o gonorrea diseminada, y pueden ocurrir como complicaciones de sífilis, tuberculosis o lepra lepromatosa. La liberación aguda de productos bacterianos durante la infección con estreptococo beta- hemolítico tipo 12 puede causar glomerulonefritis. La enfermedad suele ser autolimitada y el 95 porciento de los niños afectados se recupera en cuatro a seis semanas.
Algunos episodios temporales de artralgias agudas y erupciones cutáneas que se observan comunmente durante diversas infecciones virales pueden ser causadas por reacciones por complejos inmunes. La hepatitis crónica B o C es una causa común de glomerulonefritis, vasculitis y artritis mediada por complejos inmunes.23 Los complejos inmunes que contienen VIH causan por lo menos algunas glomerulopatías en personas VIH positivas.24
Por último, ocurre enfermedad por complejos inmunes, en especial glomerulonefritis crónica, en muchas parasitosis crónicas como paludismo, leishmaniasis, tripanosomiasis, esquistosomiasis e infecciones por filaria. En estas infecciones son comunes la estimulación crónica del sistema inmunológico por antígenos parasitarios y la síntesis policlonal de inmunoglobulinas causada por regulación defectuosa del sistema inmunológico.
Enfermedades autoinmunes
El LEG es el paradigma de las enfermedades por complejos inmunes. Produce lesiones en los riñones, vasos sanguíneos, pleura, sistema nervioso central y piel. Existe ADN en los complejos imunes renales y es posible obtener anticuerpos contra ADN de estos complejos. La frecuencia de lesiones por complejos inmunes en el LEG puede explicarse en parte por la reducción marcada en los receptores CR1 sobre los eritrocitos y los macrófagos tisulares, lo que altera el transporte de complejos inmunes al SRE. Un modelo cinético de procesamiento de los complejos inmunes sugiere que la función anormal tanto del complemento como de los receptores Fc provoca enfermedad renal activa en el LEG.25 En estudios de imagen in vivo se han demostrado varias alteraciones en el procesamiento de los complejos inmunes en pacientes con LEG, incluyendo depuración inicial acelerada de los complejos en el hígado, alteración en la retención de los complejos dentro del hígado y su liberación posterior a la circulación, y menor depuración de los complejos por el bazo.26 Pueden ocurrir alteraciones semejantes en otras enfermedades autoinmunes.
Las propiedades de los complejos inmunes circulantes en sí pueden favorecer también el depósito en el riñón. Los complejos que contienen IgA fijan menos complemento y se unen menos a los eritrocitos que los complejos que contienen IgM o IgG, por lo que se depositan en forma preferente en los riñones.27 La presencia de la subunidad A-B de C1q en los complejos inmunes correlaciona con la ocurrencia de enfermedad renal en el LEG.28 Las histonas pueden mediar el depósito glomerular de complejos ADN-antiADN de tamaño pequeño.29 Algunos sueros de pacientes con esclerosis sistémica contienen complejos inmunes que consisten en IL-6 unida a autoanticuerpos contra la citocina, y el suero de los pacientes con artritis reumatoide juvenil contiene complejos de IL-6 unidos al receptor soluble de IL-6. En ambos casos el complejo retiene la actividad de citocina que puede contribuir a la patogenia de la enfermedad.30,31
Además del depósito de complejos inmunes, otros factores pueden contribuir al daño tisular en el LEG. La observación de que el ADN de cadena simple puede unirse a la membrana basal glomerular normal sugiere que la formación de complejos ADN-anti-ADN en los riñones puede ser un tipo poco habitual de reacción inmune localizada en la que la fijación de ADN a la membrana basal glomerular es seguida de unión de anticuerpos anti-ADN. Esta reacción local puede aumentar el daño causado por el depósito de complejos inmunes preformados en los riñones. La observación de que los anticuerpos anti-ADN de pacientes con LEG con frecuencia se fijan a varios auto-antígenos aparentemente no relacionados ha originado la especulación de que los anticuerpos anti-ADN pueden dañar los órganos en forma directa al unirse a componentes aniónicos no reactivos del tejido. Esta hipótesis es motivo de controversia.32 Estudios en ratones indican que la formación de complejos inmunes en el LEG no es sino un manifestación de la disrregulación de una red de interacciones idiotipo-anti-idiotipo entre céluals B y T autorreactivas. Estos estudios destacan la importancia de los idiotipos de los autoanticuerpos en la patogenia del LEG. Los anticuerpos humanos anti-ADN comparten un número relativamente pequeño de determinantes idiotípicos, que son codificados por genes de la región variable de la cadena pesada (VH) practicamente no modificados que se conservan en la población humana.33
El ADN y sus estructuras asociadas no son los únicos antígenos presentes en los complejos inmunes en pacientes con LEG y las enfermedades autoinmunes relacionadas. El análisis sistemático de los complejos inmunes revela una mayor diversidad de la sospechda antes y sugiere que múltiples componentes de los organelos subcelulares o partículas actúan como inmunógenos in vivo. Una proporción significativa de las células B en las personas sanas producen en forma espontánea autoanticuerpos contra varias proteínas estructurales y funcionales esenciales, como proteínas de calor de choque, enzimas, tubulina, actina y colágena. Estos llamados autoantígenos pertenecen a familias de proteínas que se conservan en gran parte durante la evolución y se presentan en un rango muy amplio de organismos infecciosos. Muchos de los antígenos principales que son reconocidos en varias enfermedades infecciosas portan los mismos epítopes antigénicos contra los que reaccionan los anticuerpos humanos. Por lo tanto, las bacterias y los virus pueden iniciar algunas formas de enfermedad autoinmune y por complejos inmunes al estimular la producción de anticuerpos contra los antígenos compartidos por el agente infeccioso y el huésped humano, un proceso denominado mimetismo molecular.34
El análisis de los complejos inmunes aislados del líquido sinovial de pacientes con artritis reumatoide ha revelado reacción cruzada entre un antígeno de la cápside del virus Epstein-Barr y el fragmento Fc de la IgG humana.35 Se han encontrado otras semejanzas antigénicas en los pacientes con LEG. Alrededor del 50 porciento de los pacientes con LEG producen anticuerpos de IgG contra una proteína de 70 kd que se asocia con la ribonucleoproteína nuclear pequeña U1; esta proteína tiene una secuencia homóloga con p30ag, una proteína que se encuentra en los retrovirus de tipo C.36 Los pacientes con LEG y artritis reumatoide producen también anticuerpos contra las proteínas de calor de choque, que se encuentran en muchos organismos infecciosos;37 esta dato ha originado la especulación de que algunas enfermedades autoinmunes y por complejos de etiología desconocida puedan ser causadas por bacterias de crecimiento muy lento.38
La mayoría de los autoanticuerpos naturales son producidos por el subtipo de células B CD5+, que están controladas en forma muy estrecha por redes de células T reguladoras. Las células CD5+ constituyen solo una pequeña fracción de las células B en las personas normales, pero su número aumenta mucho en pacientes con artritis reumatoide, LEG y otras enfermedades relacionadas.
Enfermedad malignas
Se han encontrado complejos inmunes que contienen antígenos tumorales en pacientes con diversos tumores sólidos, con leucemia linfocítica aguda o crónica y con enfermedad de Hodgkin.39 En algunos casos la nefritis precede el diagnóstico clínico del trastorno maligno. Puede observarse edema angioneurótico adquirido como manifestación clínica poco habitual de la enfermedad por complejos inmunes en los pacientes con enfermedades malignas de las células B en pacientes que desarrollan anticuerpos anti-idiotipo contra el idiotipo monoclonal de las células B neoplásicas. El complejo particulado resultante activa complemento y disminuye los niveles de C1, C2, C4 e inhibidor de esterasa de C1. El tratamiento del proceso linfoproliferativo causa remisión de los episodios de angioedema.
Enfermedades gastrointestinales
La enfermedad de Crohn, la colitis ulcerativa, la cirrosis hepática, la cirrosis biliar primaria y la enfermedad celiaca pueden asociarse con lesión tisular por complejos inmunes. La menor perfusión del hígado disminuye la interacción entre los eritrocitos que tienen complejos inmunes adheridos y los fagocitos del SRE, disminuyendo su depuración fisiológica. Puede ocurrir menor depuración en pacientes con cirrosis hepática después de procedimientos quirúrgicos que desvían la sangre del hígado y durante el colapso vascular. El hígado transporta también complejos antígeno-anticuerpo de IgA de la circulación hacia la bilis. La falla de esta vía puede causar depósito de complejos de IgA en los tejidos.40
Glomerulonefritis primaria de etiología desconocida
Aparecen complejos inmunes en las siguientes enfermedades en las que no se conoce el antígeno agresor: glomerulonefritis membranosa y membranoproliferativa, enfermedad por depósitos densos, nefropatía por IgA-IgG y glomerulonefritis focal rápidamente progresiva. Se ha implicado a los autoanticuerpos contra citoplasma de neutrófilo en la patogenia de vasculitis idiopáticas como la granulomatosis de Wegener y la glomerulonefritis de medias lunas. Sin embargo, los estudios de inmunofluorescencia no suelen revelar complejos inmunes en las lesiones de pacientes con estos padecimientos.41
Deficiencias de las proteínas del complemento
La falla en la depuración de complejos inmunes dependiente del complemento explica la alta incidencia de enfermedad por complejos inmunes en pacientes con deficiencias congénitas de las proteínas de la vía clásica. En ninguno de los pacientes con deficiencia de properdina se ha observado enfermedad por complejos inmunes. Esta diferencia sugiere que la vía clásica es un mecanismo de defensa importante contra la lesión tisular mediada por complejos inmunes.3
Condiciones diversas
Se han observado depósitos de complejos inmunes en pacientes con anemia de células falciformes, nefritis asociada con púrpura de Henoch-Schönlein, dermatitis herpetiforme y crioglobulinemia mixta esencial. Existe una intensa evidencia circunstancial para un papel patogénico de los complejos inmunes en algunas formas de hipersensibilidad, en la vasculitis necrosante (púrpura palpable),18 y en la neumonía intersticial idiopática. La activación del complemento media también la vasculopatía en la dermatomiositis.42
Detección de complejos inmunes circulantes
Muchos estudios detectan complejos inmunes en el suero. La mayoría de los mismos son indirectos y se basan en una propiedad biológica o inmunoquímica del complejo. Cada ensayo reconoce en forma óptima un tipo específico de complejo inmune; por lo tanto, deben usarse varios métodos si desean detectarse todos los complejos de una muestra determinada. Algunos de los ensayos requieren que se inactive primero el complemento, lo que se hace calentando el suero a 56°C. Sin embargo, el calor puede causar resultados falsos al provocar agregación de inmunoglobulinas o al destruir algunos de los complejos.
La vigilancia estrecha de los niveles de complejos inmunes circulantes debería ayudar a evaluar el tratamiento en diversos padecimientos. Sin embargo, con frecuencia surgen dificultades al correlacionar los resultados de los complejos inmunes con otros indicadores de actividad de la enfermedad: la mayoría de los ensayos para complejos se realizan en muestras de suero, pero por lo menos una porción de los complejos que circulan in vivo se asocian con eritrocitos, y los complejos son liberados de los mismos en cierto grado durante el procesamiento de las muestras de sangre. Sin embargo, la vigilancia con ensayos de complejos inmunes estandarizados en forma cuidadosa son útiles para evaluar los resultados del tratamiento, por ejemplo, en los pacientes con nefritis lúpica. En los pacientes con infección los niveles de complejos inmunes pueden disminuir con el tratamiento exitoso, pero este cambio debe ir paralelo a una reducción en la velocidad de sedimentación globular, que es más fácil y menos costosa de medir. En los pacientes con trastornos malignos los complejos inmunes elevados pueden indicar recurrencia o progresión del crecimiento tumoral.
Debe enfatizarse que la detección de complejos inmunes en el suero no necesariamente significa que las manifestaciones clínicas de una determinada enfermedad sean causadas por daño tisular mediado por complejos. Puede requerirse el examen inmunohistoquímico de las muestras de biopsia del órgano afectado para confirmar el papel patogénico de los complejos inmunes, pero incluso este enfoque puede causar resultados equívocos. Por ejemplo, en la vasculitis mediada por complejos inmunes éstos con frecuencia son evanescentes y, por tanto, no pueden detectarse en las muestras de biopsia tomadas más de 24 horas después de la aparición clínica de la lesión.
Tratamiento
El tratamiento de la enfermedad por complejos inmunes debe dirigirse a eliminar el antígeno agresor e interrumpir la respuesta inflamatoria. Los enfoques incluyen tratar la infección subyacente o suspender la administración del medicamento sospechoso. Cuando el tamaño y otras propiedades biológicas de los complejos inmunes circulantes se modifican por un tratamiento aparentemente adecuado, las manifestaciones clínicas del daño orgánico mediado por compleos inmunes pueden exacerbarse en forma temporal. Esto ocurre, por ejemplo, cuando se pasa de un exceso de antígeno a una equivalencia antígeno-anticuerpo, lo que causa la formación de complejos inmunes más patogénicos.
Cuando la causa de la formación de los complejos inmunes no puede tratarse en forma directa, puede ser posible prevenir o minimizar el grado de daño tisular administrando fármacos antinflamatorios, esteroides y agentes citotóxicos. Los protocolos específicos de tratamiento para vasculitis, LEG y nefropatía membranosa se analizan en otros capítulos. Con excepción de los antihistamínicos, los agentes que inhiben mediadores específicos de la inflamación tisular mediada por complejos inmunes no están disponibles para uso clínico o no han sido evaluados aún en forma suficiente.
Los complejos inmunes circulantes pueden ser eliminados del suero por plasmaféresis o filtrando el plasma a través de una columna inmunoadsorbente (una resina que contiene anticuerpos contra complejos inmunes). Sin embargo, un estudio controlado ha indicado que la plasmaféresis no ofrece ventajas terapéuticas sobre el tratamiento convencional de los pacientes con nefritis lúpica severa.43 Del mismo modo, un estudio controlado sobre el uso de intercambio plasmático o leucoféresis indica que estos costosos procedimientos no son más eficaces que la aféresis ficticia para el tratamiento de la poli o dermatomiositis.44 Incluso, las transfusiones de intercambio de eritrocitos pueden no ser eficaces para eliminar los complejos libres en exceso ya que los eritrocitos transfundidos pierden con rapidez sus receptores CR1 en el paciente receptor.3
Algunas propiedades de los complejos inmunes y los procesos fisiológicos que los eliminan de la circulación pueden ser aprovechados con fines terapéuticos. Por ejemplo, la administración de gama globulina intravenosa a pacientes con trombocitopenia mediada por anticuerpos o anemia hemolítica se basa en parte en esta justificación. Al parecer los agregados de anticuerpo de la infusión de gamaglobulina compiten con los eritrocitos o plaquetas cubiertos con anticuerpo por los sitios receptores Fc en los fagocitos del SRE, esta competencia reduce la lisis de los eritrocitos o plaquetas. La gamaglobulina intravenosa modula también el daño inmune mediado por complemento.45
La mayor preocupación sobre el papel de las interleucinas y las moléculas de adhesión celular en los procesos inflamatorios ha propiciado la evaluación de tratamientos novedosos de la lesión tisular mediada por complejos inmunes en modelos animales. Los resultados iniciales son promisorios. Por ejemplo, las citocinas IL-4 e IL-10 protegen a las ratas de la lesión pulmonar inducida por complejos inmunes, lo mismo que los anticuerpos contra selectivas e integrinas que participan en la extravasación de neutrófilos y monocitos.46-48 La síntesis de NO puede ser inhibida por el análogo de L-arginina, NG-monometil-L-arginina, un agente que protege contra la lesión vascular experimental mediada por complejos inmunes.46 El hallazgo de que las cefalosporinas neutras inhiben la elastasa de leucocitos humanos sugiere que la administración de estos medicamentos relativamente no tóxicos puede beneficiar a los pacientes con enfermedad por complejos inmunes en los que la destrucción de la matriz extracelular de los tejidos es la característica dominante.49,50
Un dato interesante es que la mayoría de los pacientes tratados con metildopa que desarrollan autoanticuerpos de IgG contra los eritrocitos no sufren de anemia hemolítica. La investigación de esta aparente discrepancia sugiere que la metildopa inhibe las funciones Fc-dependientes del SRE.51 Este dato justifica la investigación sobre el uso de metildopa para tratar los casos de citopenia causados por pérdida excesiva de los eritrocitos cubiertos por anticuerpos.
Reconocimientos
Figuras 1 y 3 a 6 Dana Burns-Pfizer.
Figura 2 George Kelvin.
Figura 7 Al Miller.
Bibliografía
DR. WILLY F. PIESSENS
El término enfermedad por complejos inmunes se refiere a un grupo de padecimientos cuya patogenia incluye el daño tisular por reacciones de complejos inmunes. Debido a que siempre que los anticuerpos interactúan con antígenos exógenos o endógenos se forman complejos inmunes potencialmente dañinos, las enfermedades por complejos inmunes son heterogéneas, con causas y manifestaciones clínicas muy diversas.
Fisiología o fisiopatología
FORMACION DE UNA RED ANTIGENO-ANTICUERPO
Varios factores determinan el tamaño y solubilidad de un complejo inmune formado in vitro, incluyendo la relación molar de anticuerpo a antígeno, las propiedades intrínsecas de estas dos moléculas y el medio en que la reacción tiene lugar. Los estudios clásicos sobre reacciones de inmunoprecipitación ilustran de la mejor manera la contribución de las cantidades relativas de antígeno y anticuerpo al tamaño del complejo. Los complejos más grandes, que son los menos solubles, se forman cuando la relación molar de anticuerpo a antígeno se aproxima a 1 [ver figura 1]. Esta región se conoce como zona de equivalencia, en la que no puede detectarse ni antígeno ni anticuerpo libre en el sobrenadante de la reacción de precipitación. Los complejos que se forman cuando existe exceso de antígeno o de anticuerpo son más pequeños que los formados en la zona de equivalencia y causan precipitados de menor tamaño [ver figura 2].
|
|
El complemento es otro determinante importante del tamaño de un complejo inmune.2 La interacción de un anticuerpo IgM o IgG fijador de complemento con un antígeno induce cambios conformacionales en la porción Fc de la molécula de inmunoglobulina que desencadenan la activación del complemento a través de la vía clásica. La unión de C1 a la inmunoglobulina, que inicia la reacción del complemento, retrasa las interacciones Fc-Fc y en circunstancias normales provoca la formación de complejos que son menores que los que se formarían en ausencia de complemento [ver figura 3]. Los complejos que contienen IgA, una clase de anticuerpos que no fijan C1, pueden activar la vía alterna del complemento, lo que ocasiona depósito de C3b. LA molécula C3b debilita las fuerzas que mantienen juntos al antígeno y al anticuerpo. El grado de fijación varía con el tamaño del complejo (los complejos más grandes fijan más complemento que los pequeños) y con el isotipo del anticuerpo (IgM>IgG>IgA). Por lo general los complejos que contienen IgE no fijan complemento.
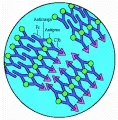
|
| Figura 3 |
| Red de complejos inmunes |
DEPURACION FISIOLOGICA DE LOS COMPLEJOS INMUNES
En condiciones fisiológicas los complejos inmunes que se forman directamente dentro de los tejidos son transportados por los linfáticos para drenar en los ganglios linfáticos,3 mientras que los complejos de la sangre son eliminados de la circulación por el sistema reticuloendotelial (SRE). Los fagocitos mononucleares en los ganglios linfáticos, bazo e hígado fagocitan y degradan los complejos.
La opsonización de los complejos inmunes (la unión covalente de muchas moléculas C3b al complejo antígeno-anticuerpo cuando se activa complemento por la vía clásica) facilita la depuración de los complejos inmunes de la circulación. La opsonización permite que las células que poseen un receptor específico para C3b, el receptor 1 de complemento (CR1, por sus siglas en inglés, n. del t.), fijen a los complejos opsonizados (cubiertos por C3b) y los entreguen al SRE.2,4 El CR1 está presente en muchos tipos de células, pero más del 90 porciento de los CR1 en la sangre que encuentran en las membranas de los eritrocitos. Por lo tanto, los eritrocitos son los principales transportadores de complejos inmunes al SRE.5 Este modo de transporte parece proteger al endotelio vascular del daño mediado por complejos inmunes.
Varios factores facilitan la transferencia de complejos inmunes de los eritrocitos a los fagocitos del SRE en el bazo y el hígado. La activación de la vía alterna del complemento induce la liberación de los complejos unidos a los eritrocitos, la unión de los complejos inmunes a los eritrocitos se debilita cuando el factor I del complemento en el plasma separa al C3b unido al complejo. Los fagocitos poseen más receptors CR1 que los eritrocitos. Además, los fagocitos contienen receptores para la porción Fc de la IgG, que no existen en los eritrocitos [ver figura 4]. Los receptores Fc en las células del SRE no solo capturan los complejos inmunes unidos a los eritrocitos, sino también eliminan a los complejos inmunes que contienen IgG libres de la circulación.6 Los complejos grandes y opsonizados son eliminados en forma eficiente de la sangre en minutos. Los complejos pequeños son malos activadores del complemento y pueden continuar circulando durante muchos días.
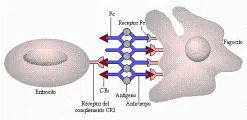
|
| Figura 4 |
| Depuración de complejos inmunes |
DEPOSITO ABERRANTE DE LOS COMPLEJOS INMUNES
Las alteraciones en la depuración fisiológica de los complejos inmunes pueden causar depósitos aberrantes en los vasos sanguíneos y los tejidos circundantes. Varios procesos estimulan la liberación local de sustancias vasoactivas que aumentan la permeabilidad vascular. Los antígenos en los complejos circulantes pueden interactuar con anticuerpos IgE específicos y anticuerpos acoplados con receptores Fc de los basófilos circulantes , provocando la liberación de sustancias como histamina y factor activador de plaquetas (FAP). El FAP, las anafilotoxinas C3a y C5a generadas durante la activación del complemento o los complejos inmunes per se causan agregación y degranulación plaquetaria, con liberación de histamina y serotonina. Todos estos factores facilitan el depósito de complejos inmunes en las paredes de los vasos sanguíneos y migración de los complejos hacia los tejidos. Estas sustancias vasoactivas también provocan la urticaria que ocurre en la enfermedad por complejos inmunes.
Los depósitos pueden ocurrir en cualquier parte del organismo, pero las lesiones tienden a localizarse en áreas en donde las plaquetas se adhieren con más frecuencia al endotelio por la mayor presión sanguínea y la turbulencia. Varios factores adicionales pueden explicar porqué algunos órganos, como el riñón, se afectan con más frecuencia que otros.7 La combinación de flujo sanguíneo rápido, filtración intensa y presión sanguínea alta en los glomérulos renales facilita el depósito de complejos inmunes. Los receptores para los componentes activados del complemento en los podocitos de los glomérulos normales pueden causar que los complejos inmunes se concentren en este sitio.8 Estos receptores están también presentes en el plexo coroideo, otro sitio común de depósitos de complejos inmunes. Los receptores para los componentes activados del complemento en los podocitos se concentran en este sitio.8 Estos receptores están presentes también en el plexo coroideo, otro sitio común de depósito de complejos inmunes. Los complejos son atrapados con facilidad en la membrana basal glomerular porque el endotlio vascular renal es fenestrado (muy poroso) y el intersticio renal contiene receptores para Fc. Además, las membranas basales, se encuentren en el glomérulo renal o en la piel, tienen carga negativa, por lo que retienen los complejos inmunes con carga positiva por más tiempo que los neutros o con carga negativa. El efecto de la carga puede explicar el depósito frecuente de complejos inmunes en las regiones subendoteliales del glomérulo y en la unión dermoepidérmica de la piel.
Cuando la capacidad de los eritrocitos para actuar como transportadores de los complejos inmunes está alterada, los complejos se depositan con más facilidad en los vasos sanguíneos y no son eliminados por los fagocitos mononucleares del bazo y el hígado. Esta alteración ocurre en los pacientes que tienen lupus eritematoso generalizado (LEG), un padecimiento en el que los eritrocitos tienen menor número de receptores C3b.9 De igual modo, cuando los fagocitos en el hígado y el bazo están disminuidos en número, bloqueados por haber captado antes una partícula, o deficientes en complemento o receptores Fc, se depositan complejos dañinos en otros órganos. La deficiencia del receptor C3b se ha asociado con vasculitis inmune y paniculitis en la piel de pacientes con LEG.10
La actividad funcional de los receptores Fc sobre las células del SRE puede evaluarse por medio de eritrocitos autólogos radiomarcados y cubiertos con anticuerpos IgG anti-eritrocito in vitro. Cuando se inyectan estás células al paciente, la velocidad de desaparición de las mismas correlaciona con la actividad del receptor Fc. Los estudios de pacientes con LEG muestran un defecto importante en la depuración específica del receptor Fc que correlaciona con la actividad de la enfermedad.11 La función Fc de los macrófagos también está alterada en pacientes con nefropatía terminal.12
LESION TISULAR MEDIADA POR COMPLEJOS INMUNES: REACCION DE ARTHUS
La reacción de Arthus es un proceso hemorrágico y necrótico agudo que ocurre en dos a cuatro horas después de que se inyecta el antígeno en la piel u otros tejidos de un animal inmunizado. Esta reacción se considera el prototipo de la lesión tisular aguda y localizada mediada por complejos inmunes, y se ha usado para estudiar la secuencia de eventos por los que el depósito de complejos inmunes provoca daño tisular. Al inicio se desarrolla una intensa infiltración de leucocitos polimorfonucleares dentro y alrededor de los vasos sanguíneos. La alteración de la pared vascular causa que los eritrocitos salgan hacia los tejidos. Posteriormente linfocitos, células plasmáticas y monocitos infiltran las lesiones y ocurre proliferación de las células endoteliales. Puede detectarse anticuerpo, antígeno y complemento en las lesiones por medio de microscopía inmunofluorescente [ver figura 5]. Los estudios de esta reacción indican que los componentes del complemento y los neutrófilos son esenciales para la formación de estas lesiones.
|
|


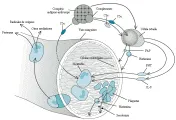
|
| Figura 6 |
| Patogenia de la enfermedad por complejos inmunes |
C3b, que se forma cuando los complejos inmunes activan complemento, se fija con rapidez a la membrana basal de los vasos. La fijación de los neutrófilos a las estructuras cubiertas por C3b a través del CR1 causa también liberación de gránulos lisosómicos en ausencia de fagocitosis. Por último, C3e, un fragmento derivado de la mayor degradación de C3b, estimula la neutrofilia reactiva al liberar células de la médula ósea.
C5a, así como citocinas como la interleucina-8 (IL-8) y el factor de necrosis tumoral-alfa (FNT-alfa), liberados por las células cebadas, son potentes quimiotácticos para los neutrófilos. Estas células se acumulan en el sitio de reacción, fijan complejos antígeno-anticuerpo a través de sus receptores Fc y fagocitan los complejos. Durante este proceso los neutrófilos liberan un amplio rango de productos que pueden dañar los componentes tisulares. Estos productos incluyen moléculas oxidantes y radicales como el H2O2 y los aniones superóxido, péptidos catiónicos que aumentan la permeabilidad capilar, causan contracción del músculo liso y desecadenan mayor liberación de los mediadores de células cebadas, y enzimas que degradan las membranas basales en los riñones y vasos sanguíneos, glucosaminoglicanos y cartílago en las articulaciones, fibras elásticas en los vasos sanguíneos y colágena en muchos tejidos. La liberación de estas hidrolasas puede ser inhibida en parte por agentes que aumentan el monofosfato cíclico de adenosina (AMPc), como la prostaglandina E1. La lesión tisular causada por el depósito de complejos inmunes también está mediada en parte por exceso de óxido nítrico liberado en forma local (NO), un mediador potente y pleiotrópico secretado por macrófagos, neutrófilos y células endoteliales activados. El FAP induce sintetasa de óxido nítrico, una enzima que genera NO.13
El modelo tradicional sostiene que la reacción de Arthus inicia cuando los complejos inmunes activan al complemento en los tejidos a través de la vía clásica. Se generan fragmentos quimiotácticos del complmento para atraer neutrófilos y estimular a éstas células y a las células cebadas a liberar diversos productos solubles que causan daño tisular. Un estudio reciente pone en duda la secuencia de los eventos. Los resultados del estudio sugieren que la reacción de Arthus puede iniciarse por la interacción de los complejos inmunes con receptores Fc de las células y ser después amplificada por productos activados del complemento y mediadores celulares.14 Otros investigadores destacan la contribución de las citocinas quimiotácticas y las moléculas de adhesión en la extravasación y activación de neutrófilos15 e indican que el antagonista del receptor de la interleucina-1 (IL-1) modula el daño tisular mediado por complejos inmunes.16 Estas observaciones han originado nuevos enfoques de tratamiento de los síndromes de complejos inmunes en modelos animales [ver adelante, Tratamiento].
OTROS EFECTOS DE LOS COMPLEJOS INMUNES
En condiciones fisiológicas los complejos antígeno-anticuerpo tienen funciones inmunorreguladoras importantes. El antígeno es transportado a través de la linfa hacia los centros germinales de los ganglios linfáticos regionales, en donde forma un complejo con anticuerpo y complemento. La retención del antígeno en los centros germinales es necesaria para el desarrollo de células B de memoria. Las interacciones idiotipo-antidiotipo, un tipo especializado de formación de complejo inmune, parecen ser reguladores generales de las respuestas inmunes. Los complejos inmunes pueden inducir también la activación o la tolerancia a inyecciones subsecuentes de antígeno solo, un hecho relevante para la programación de las vacunas.17
La interacción dependiente de Fc o de complemento de las células con blancos específicos puede ser inhibida por complejos inmunes que compiten por los mismos receptores. Esta inhibición, o bloqueo, parece particiar en el escape de las células tumorales y parásitos de la citotoxicidad mediada por células dependientes de anticuerpos. En el suero de los pacientes con fibrosis quística existen niveles altos de complejos inmunes que contienen IgG2 y que bloquean los receptores Fc y así inhiben la fagocitosis de Pseudomonas aeruginosa. Los complejos inmunes en el suero de estos pacientes pueden activar también células T para destruir células B que producen anticuerpos opsonizantes específicos. Estos dos mecanismos pueden ser responsables de la infección crónica por Pseudomonas que se presenta en las vías respiratorias en la fibrosis quística.18,19
Enfermedad por complejos inmunes localizada
Los antígenos que desencadenan la enfermedad por complejos inmunes localizada pueden ser endógenos estructurales, celulares, autoantígenos tisulares secretados, o antígenos exógenos que se concentran en ciertos órganos [ver tabla 1]. Los síntomas clínicos que ocasionan varían de acuerdo con la localización de la reacción por complejo inmune. El complemento puede tener un papel muy importante en la patogenia de algunas enfermedades por complejos inmunes localizadas, como lo ilustran los estudios experimentales sobre miastenia gravis. En este modelo los anticuerpos reaccionan con receptores de acetilcolina, pero la destrucción inflamatoria de las placas motoras terminales no ocurre cuando se evita la activación del complemento.
|
|||||||||||||||||||||||||
|
Por lo general no ocurre una reacción de Arthus cuando se inyecta a una persona un antígeno extraño porque no existe suficiente anticuerpo circulante contra el antígeno. Sin embargo, las concentraciones locales de antígeno en una persona sensibilizada que tiene anticuerpos específicos pueden causar lesión tisular mediada por complejos inmunes [ver tabla 1]. La inflamación que se desarrolla después de una inyección de refuerzo de toxoide tetánico puede ser en parte una reacción de tipo Arthus. Cuando las personas que tienen anticuerpos específicos contra diferentes antígenos micóticos o proteínas animales inhalan estos antígenos, las reacciones tipo Arthus en el pulmón causan neumonitis por hipersensibilidad. Los síntomas consisten en fiebre, tos y disnea, que comienzan cuatro a seis horas después de la exposición al antígeno específico, alcanzan su máximo después de 12 a 24 horas y suelen resolverse en dos a tres días. Las radiografías de los pulmones durante la fase aguda muestran edema intersticialy, en ocasiones, infiltrados en parches. La biopsia pulmonar revela infiltración por granulocitos y linfocitos. En la enfermedad pulmonar crónica por complejos inmunes se observan los cambios típicos del enfisema (infiltración con macrófagos y pérdida de las fibras de elastina y colágena). La enfermedad por complejos inmunes de los pulmones también ocurre en niños que han recibido vacunas por virus muertos que inducen formación de anticuerpos pero no producen inmunidad protectora, como la vacuna del sarampión con virus muertos. Cuando estos niños son expuestos en una etapa posterior a virus vivos los antígenos se combinan con los anticuerpos y producen lesión por complejos inmunes.
Enfermedad sistémica por complejos inmunes
ENFERMEDAD DEL SUERO
La enfermedad del suero fue descrita por primera vez a principios del siglo XX en pacientes que recibieron suero de caballo para el tratamiento de enfermedades infecciosas. La enfermedad del suero es común en pacientes que reciben inmunoglobulina antitimocítica equina como tratamiento para la falla de la médula ósea. También ocurre en pacientes infectados por el virus de inmunodeficiencia humana (VIH) que desarrollan anticuerpos contra suero fetal de ternera después de infusiones de linfocitos singénicos20 y en algunos pacientes tratados con estreptocinasa.21
Varios fármacos no proteináceos pueden provocar enfermedad del suero, incluyendo penicilina, oro, penicilamina, sulfonamidas, tiouracilos, hidantoínas, tiacidas, fenilbutazona, ácido aminosalicílico y estreptomicina. Estos medicamentos parecen actuar como haptenos que inducen respuestas de anticuerpos cuando se acoplan con proteínas transportadoras. La administración repetida de proteínas extrañas y fármacos de depósito o vida media muy larga, como penicilamina y oro, puede inducir enfermedad del suero crónica.
Típicamente, ocho a 12 días después de la administración de una gran cantidad de suero de especies extrañas el paciente muestra fiebre, malestar, ganglios linfáticos aumentados de tamaño y dolorosos, articulaciones dolorosas e inflamadas, erupciones cutáneas, molestias gastrointestinales y crecimiento del bazo. Los estudios de laboratorio pueden mostrar leucocitosis, proteinuria, menor actividad del complemento sérico y, en ocasiones, eosinofilia. Los síntomas suelen desaparecer después de una semana, momento en el que pueden detectarse anticuerpos contra los antígenos extraños en el suero. El inicio de los síntomas ocurre en forma más temprana después de una segunda inyección de suero. la enfermedad del suero suele ser benigna y autolimitada, sus principales complicaciones son vasculitis, glomerulonefritis, artritis y neuritis. En ocasiones ocurre síndrome de Guillain-Barré. Aunque los signos y síntomas neurológicos pueden durar un tiempo prolongado, la recuperación suele ser completa.
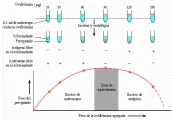
La patogenia de la enfermedad del suero en humanos es semejante a la descrita en modelos animales [ver figura 7].22 La enfermedad clínica coincide con el desarrollo de niveles muy altos de complejos inmunes circulantes y disminuye en forma importante los niveles séricos de C3, C4 y actividad del complemento hemolítico. Pueden detectarse inmunoglobulinas y C3 por microscopía con inmunofluorescencia en las paredes de los vasos cutáneos pequeños en la mayoría de los pacientes con enfermedad del suero aguda. Una banda serpiginosa no habitual de eritema en las palmas y plantas puede ser el primer signo de esta entidad.

|
| Figura 7 |
| Enfermedad del suero experimental |
NEFRITIS CRONICA POR COMPLEJOS INMUNES
Debido a que los glomérulos renales son especialmente vulnerables a la lesión inmune mediada por complejos, la glomerulonefritis es una característica frecuente de muchas enfermedades por complejos inmunes y con frecuencia ocurre en ausencia de otras manifestaciones clínicas. Los complejos formados en la circulación suelen depositarse en las áreas mesangiales y endoteliales y producir un patrón irregular típico en la inmunofluorescencia ('lumpy-bumpy').
OTRAS ENFERMEDADES POR COMPLEJOS INMUNES
Infecciones
La glomerulonefritis y otras manifestaciones de vasculitis pueden ocurrir en asociación con muchas infecciones bacterianas persistentes (v.gr., endocarditis bacteriana subaguda), infecciones de derivaciones arteriovenosas o gonorrea diseminada, y pueden ocurrir como complicaciones de sífilis, tuberculosis o lepra lepromatosa. La liberación aguda de productos bacterianos durante la infección con estreptococo beta- hemolítico tipo 12 puede causar glomerulonefritis. La enfermedad suele ser autolimitada y el 95 porciento de los niños afectados se recupera en cuatro a seis semanas.
Algunos episodios temporales de artralgias agudas y erupciones cutáneas que se observan comunmente durante diversas infecciones virales pueden ser causadas por reacciones por complejos inmunes. La hepatitis crónica B o C es una causa común de glomerulonefritis, vasculitis y artritis mediada por complejos inmunes.23 Los complejos inmunes que contienen VIH causan por lo menos algunas glomerulopatías en personas VIH positivas.24
Por último, ocurre enfermedad por complejos inmunes, en especial glomerulonefritis crónica, en muchas parasitosis crónicas como paludismo, leishmaniasis, tripanosomiasis, esquistosomiasis e infecciones por filaria. En estas infecciones son comunes la estimulación crónica del sistema inmunológico por antígenos parasitarios y la síntesis policlonal de inmunoglobulinas causada por regulación defectuosa del sistema inmunológico.
Enfermedades autoinmunes
El LEG es el paradigma de las enfermedades por complejos inmunes. Produce lesiones en los riñones, vasos sanguíneos, pleura, sistema nervioso central y piel. Existe ADN en los complejos imunes renales y es posible obtener anticuerpos contra ADN de estos complejos. La frecuencia de lesiones por complejos inmunes en el LEG puede explicarse en parte por la reducción marcada en los receptores CR1 sobre los eritrocitos y los macrófagos tisulares, lo que altera el transporte de complejos inmunes al SRE. Un modelo cinético de procesamiento de los complejos inmunes sugiere que la función anormal tanto del complemento como de los receptores Fc provoca enfermedad renal activa en el LEG.25 En estudios de imagen in vivo se han demostrado varias alteraciones en el procesamiento de los complejos inmunes en pacientes con LEG, incluyendo depuración inicial acelerada de los complejos en el hígado, alteración en la retención de los complejos dentro del hígado y su liberación posterior a la circulación, y menor depuración de los complejos por el bazo.26 Pueden ocurrir alteraciones semejantes en otras enfermedades autoinmunes.
Las propiedades de los complejos inmunes circulantes en sí pueden favorecer también el depósito en el riñón. Los complejos que contienen IgA fijan menos complemento y se unen menos a los eritrocitos que los complejos que contienen IgM o IgG, por lo que se depositan en forma preferente en los riñones.27 La presencia de la subunidad A-B de C1q en los complejos inmunes correlaciona con la ocurrencia de enfermedad renal en el LEG.28 Las histonas pueden mediar el depósito glomerular de complejos ADN-antiADN de tamaño pequeño.29 Algunos sueros de pacientes con esclerosis sistémica contienen complejos inmunes que consisten en IL-6 unida a autoanticuerpos contra la citocina, y el suero de los pacientes con artritis reumatoide juvenil contiene complejos de IL-6 unidos al receptor soluble de IL-6. En ambos casos el complejo retiene la actividad de citocina que puede contribuir a la patogenia de la enfermedad.30,31
Además del depósito de complejos inmunes, otros factores pueden contribuir al daño tisular en el LEG. La observación de que el ADN de cadena simple puede unirse a la membrana basal glomerular normal sugiere que la formación de complejos ADN-anti-ADN en los riñones puede ser un tipo poco habitual de reacción inmune localizada en la que la fijación de ADN a la membrana basal glomerular es seguida de unión de anticuerpos anti-ADN. Esta reacción local puede aumentar el daño causado por el depósito de complejos inmunes preformados en los riñones. La observación de que los anticuerpos anti-ADN de pacientes con LEG con frecuencia se fijan a varios auto-antígenos aparentemente no relacionados ha originado la especulación de que los anticuerpos anti-ADN pueden dañar los órganos en forma directa al unirse a componentes aniónicos no reactivos del tejido. Esta hipótesis es motivo de controversia.32 Estudios en ratones indican que la formación de complejos inmunes en el LEG no es sino un manifestación de la disrregulación de una red de interacciones idiotipo-anti-idiotipo entre céluals B y T autorreactivas. Estos estudios destacan la importancia de los idiotipos de los autoanticuerpos en la patogenia del LEG. Los anticuerpos humanos anti-ADN comparten un número relativamente pequeño de determinantes idiotípicos, que son codificados por genes de la región variable de la cadena pesada (VH) practicamente no modificados que se conservan en la población humana.33
El ADN y sus estructuras asociadas no son los únicos antígenos presentes en los complejos inmunes en pacientes con LEG y las enfermedades autoinmunes relacionadas. El análisis sistemático de los complejos inmunes revela una mayor diversidad de la sospechda antes y sugiere que múltiples componentes de los organelos subcelulares o partículas actúan como inmunógenos in vivo. Una proporción significativa de las células B en las personas sanas producen en forma espontánea autoanticuerpos contra varias proteínas estructurales y funcionales esenciales, como proteínas de calor de choque, enzimas, tubulina, actina y colágena. Estos llamados autoantígenos pertenecen a familias de proteínas que se conservan en gran parte durante la evolución y se presentan en un rango muy amplio de organismos infecciosos. Muchos de los antígenos principales que son reconocidos en varias enfermedades infecciosas portan los mismos epítopes antigénicos contra los que reaccionan los anticuerpos humanos. Por lo tanto, las bacterias y los virus pueden iniciar algunas formas de enfermedad autoinmune y por complejos inmunes al estimular la producción de anticuerpos contra los antígenos compartidos por el agente infeccioso y el huésped humano, un proceso denominado mimetismo molecular.34
El análisis de los complejos inmunes aislados del líquido sinovial de pacientes con artritis reumatoide ha revelado reacción cruzada entre un antígeno de la cápside del virus Epstein-Barr y el fragmento Fc de la IgG humana.35 Se han encontrado otras semejanzas antigénicas en los pacientes con LEG. Alrededor del 50 porciento de los pacientes con LEG producen anticuerpos de IgG contra una proteína de 70 kd que se asocia con la ribonucleoproteína nuclear pequeña U1; esta proteína tiene una secuencia homóloga con p30ag, una proteína que se encuentra en los retrovirus de tipo C.36 Los pacientes con LEG y artritis reumatoide producen también anticuerpos contra las proteínas de calor de choque, que se encuentran en muchos organismos infecciosos;37 esta dato ha originado la especulación de que algunas enfermedades autoinmunes y por complejos de etiología desconocida puedan ser causadas por bacterias de crecimiento muy lento.38
La mayoría de los autoanticuerpos naturales son producidos por el subtipo de células B CD5+, que están controladas en forma muy estrecha por redes de células T reguladoras. Las células CD5+ constituyen solo una pequeña fracción de las células B en las personas normales, pero su número aumenta mucho en pacientes con artritis reumatoide, LEG y otras enfermedades relacionadas.
Enfermedad malignas
Se han encontrado complejos inmunes que contienen antígenos tumorales en pacientes con diversos tumores sólidos, con leucemia linfocítica aguda o crónica y con enfermedad de Hodgkin.39 En algunos casos la nefritis precede el diagnóstico clínico del trastorno maligno. Puede observarse edema angioneurótico adquirido como manifestación clínica poco habitual de la enfermedad por complejos inmunes en los pacientes con enfermedades malignas de las células B en pacientes que desarrollan anticuerpos anti-idiotipo contra el idiotipo monoclonal de las células B neoplásicas. El complejo particulado resultante activa complemento y disminuye los niveles de C1, C2, C4 e inhibidor de esterasa de C1. El tratamiento del proceso linfoproliferativo causa remisión de los episodios de angioedema.
Enfermedades gastrointestinales
La enfermedad de Crohn, la colitis ulcerativa, la cirrosis hepática, la cirrosis biliar primaria y la enfermedad celiaca pueden asociarse con lesión tisular por complejos inmunes. La menor perfusión del hígado disminuye la interacción entre los eritrocitos que tienen complejos inmunes adheridos y los fagocitos del SRE, disminuyendo su depuración fisiológica. Puede ocurrir menor depuración en pacientes con cirrosis hepática después de procedimientos quirúrgicos que desvían la sangre del hígado y durante el colapso vascular. El hígado transporta también complejos antígeno-anticuerpo de IgA de la circulación hacia la bilis. La falla de esta vía puede causar depósito de complejos de IgA en los tejidos.40
Glomerulonefritis primaria de etiología desconocida
Aparecen complejos inmunes en las siguientes enfermedades en las que no se conoce el antígeno agresor: glomerulonefritis membranosa y membranoproliferativa, enfermedad por depósitos densos, nefropatía por IgA-IgG y glomerulonefritis focal rápidamente progresiva. Se ha implicado a los autoanticuerpos contra citoplasma de neutrófilo en la patogenia de vasculitis idiopáticas como la granulomatosis de Wegener y la glomerulonefritis de medias lunas. Sin embargo, los estudios de inmunofluorescencia no suelen revelar complejos inmunes en las lesiones de pacientes con estos padecimientos.41
Deficiencias de las proteínas del complemento
La falla en la depuración de complejos inmunes dependiente del complemento explica la alta incidencia de enfermedad por complejos inmunes en pacientes con deficiencias congénitas de las proteínas de la vía clásica. En ninguno de los pacientes con deficiencia de properdina se ha observado enfermedad por complejos inmunes. Esta diferencia sugiere que la vía clásica es un mecanismo de defensa importante contra la lesión tisular mediada por complejos inmunes.3
Condiciones diversas
Se han observado depósitos de complejos inmunes en pacientes con anemia de células falciformes, nefritis asociada con púrpura de Henoch-Schönlein, dermatitis herpetiforme y crioglobulinemia mixta esencial. Existe una intensa evidencia circunstancial para un papel patogénico de los complejos inmunes en algunas formas de hipersensibilidad, en la vasculitis necrosante (púrpura palpable),18 y en la neumonía intersticial idiopática. La activación del complemento media también la vasculopatía en la dermatomiositis.42
Detección de complejos inmunes circulantes
Muchos estudios detectan complejos inmunes en el suero. La mayoría de los mismos son indirectos y se basan en una propiedad biológica o inmunoquímica del complejo. Cada ensayo reconoce en forma óptima un tipo específico de complejo inmune; por lo tanto, deben usarse varios métodos si desean detectarse todos los complejos de una muestra determinada. Algunos de los ensayos requieren que se inactive primero el complemento, lo que se hace calentando el suero a 56°C. Sin embargo, el calor puede causar resultados falsos al provocar agregación de inmunoglobulinas o al destruir algunos de los complejos.
La vigilancia estrecha de los niveles de complejos inmunes circulantes debería ayudar a evaluar el tratamiento en diversos padecimientos. Sin embargo, con frecuencia surgen dificultades al correlacionar los resultados de los complejos inmunes con otros indicadores de actividad de la enfermedad: la mayoría de los ensayos para complejos se realizan en muestras de suero, pero por lo menos una porción de los complejos que circulan in vivo se asocian con eritrocitos, y los complejos son liberados de los mismos en cierto grado durante el procesamiento de las muestras de sangre. Sin embargo, la vigilancia con ensayos de complejos inmunes estandarizados en forma cuidadosa son útiles para evaluar los resultados del tratamiento, por ejemplo, en los pacientes con nefritis lúpica. En los pacientes con infección los niveles de complejos inmunes pueden disminuir con el tratamiento exitoso, pero este cambio debe ir paralelo a una reducción en la velocidad de sedimentación globular, que es más fácil y menos costosa de medir. En los pacientes con trastornos malignos los complejos inmunes elevados pueden indicar recurrencia o progresión del crecimiento tumoral.
Debe enfatizarse que la detección de complejos inmunes en el suero no necesariamente significa que las manifestaciones clínicas de una determinada enfermedad sean causadas por daño tisular mediado por complejos. Puede requerirse el examen inmunohistoquímico de las muestras de biopsia del órgano afectado para confirmar el papel patogénico de los complejos inmunes, pero incluso este enfoque puede causar resultados equívocos. Por ejemplo, en la vasculitis mediada por complejos inmunes éstos con frecuencia son evanescentes y, por tanto, no pueden detectarse en las muestras de biopsia tomadas más de 24 horas después de la aparición clínica de la lesión.
Tratamiento
El tratamiento de la enfermedad por complejos inmunes debe dirigirse a eliminar el antígeno agresor e interrumpir la respuesta inflamatoria. Los enfoques incluyen tratar la infección subyacente o suspender la administración del medicamento sospechoso. Cuando el tamaño y otras propiedades biológicas de los complejos inmunes circulantes se modifican por un tratamiento aparentemente adecuado, las manifestaciones clínicas del daño orgánico mediado por compleos inmunes pueden exacerbarse en forma temporal. Esto ocurre, por ejemplo, cuando se pasa de un exceso de antígeno a una equivalencia antígeno-anticuerpo, lo que causa la formación de complejos inmunes más patogénicos.
Cuando la causa de la formación de los complejos inmunes no puede tratarse en forma directa, puede ser posible prevenir o minimizar el grado de daño tisular administrando fármacos antinflamatorios, esteroides y agentes citotóxicos. Los protocolos específicos de tratamiento para vasculitis, LEG y nefropatía membranosa se analizan en otros capítulos. Con excepción de los antihistamínicos, los agentes que inhiben mediadores específicos de la inflamación tisular mediada por complejos inmunes no están disponibles para uso clínico o no han sido evaluados aún en forma suficiente.
Los complejos inmunes circulantes pueden ser eliminados del suero por plasmaféresis o filtrando el plasma a través de una columna inmunoadsorbente (una resina que contiene anticuerpos contra complejos inmunes). Sin embargo, un estudio controlado ha indicado que la plasmaféresis no ofrece ventajas terapéuticas sobre el tratamiento convencional de los pacientes con nefritis lúpica severa.43 Del mismo modo, un estudio controlado sobre el uso de intercambio plasmático o leucoféresis indica que estos costosos procedimientos no son más eficaces que la aféresis ficticia para el tratamiento de la poli o dermatomiositis.44 Incluso, las transfusiones de intercambio de eritrocitos pueden no ser eficaces para eliminar los complejos libres en exceso ya que los eritrocitos transfundidos pierden con rapidez sus receptores CR1 en el paciente receptor.3
Algunas propiedades de los complejos inmunes y los procesos fisiológicos que los eliminan de la circulación pueden ser aprovechados con fines terapéuticos. Por ejemplo, la administración de gama globulina intravenosa a pacientes con trombocitopenia mediada por anticuerpos o anemia hemolítica se basa en parte en esta justificación. Al parecer los agregados de anticuerpo de la infusión de gamaglobulina compiten con los eritrocitos o plaquetas cubiertos con anticuerpo por los sitios receptores Fc en los fagocitos del SRE, esta competencia reduce la lisis de los eritrocitos o plaquetas. La gamaglobulina intravenosa modula también el daño inmune mediado por complemento.45
La mayor preocupación sobre el papel de las interleucinas y las moléculas de adhesión celular en los procesos inflamatorios ha propiciado la evaluación de tratamientos novedosos de la lesión tisular mediada por complejos inmunes en modelos animales. Los resultados iniciales son promisorios. Por ejemplo, las citocinas IL-4 e IL-10 protegen a las ratas de la lesión pulmonar inducida por complejos inmunes, lo mismo que los anticuerpos contra selectivas e integrinas que participan en la extravasación de neutrófilos y monocitos.46-48 La síntesis de NO puede ser inhibida por el análogo de L-arginina, NG-monometil-L-arginina, un agente que protege contra la lesión vascular experimental mediada por complejos inmunes.46 El hallazgo de que las cefalosporinas neutras inhiben la elastasa de leucocitos humanos sugiere que la administración de estos medicamentos relativamente no tóxicos puede beneficiar a los pacientes con enfermedad por complejos inmunes en los que la destrucción de la matriz extracelular de los tejidos es la característica dominante.49,50
Un dato interesante es que la mayoría de los pacientes tratados con metildopa que desarrollan autoanticuerpos de IgG contra los eritrocitos no sufren de anemia hemolítica. La investigación de esta aparente discrepancia sugiere que la metildopa inhibe las funciones Fc-dependientes del SRE.51 Este dato justifica la investigación sobre el uso de metildopa para tratar los casos de citopenia causados por pérdida excesiva de los eritrocitos cubiertos por anticuerpos.
Figuras 1 y 3 a 6 Dana Burns-Pfizer.
Figura 2 George Kelvin.
Figura 7 Al Miller.
Bibliografía
-
Mannik M: Physiochemical and functional relationships of
immune complexes. J Invest Dermatol 74:333, 1980
-
Schifferli JA, Ng YC, Peters DK: The role of complement and
its receptor in the elimination of immune complexes. N Engl J Med 315:488,
1986 [PMID
2942776]
-
Van den Berg TK, Yoshida K, Dijkstra CD: Mechanism of immune
complex trapping by follicular dendritic cells. Curr Top Microbiol Immunol
201:49, 1995 [PMID
7587352]
-
Davies KA, Erlendsson K, Beynon HL, et al: Splenic uptake
of immune complexes in man is complement-dependent. J Immunol 151:3866,
1993 [PMID
8376807]
-
Nielsen CH, Matthiesen SH, Lyng I, et al: The role of complement
receptor type 1 (CR1, CD35) in determining the cellular distribution of
opsonized immune complexes between whole blood cells: kinetic analysis
of the buffering capacity of erythrocytes. Immunology 90:129, 1997 [PMID
9038723]
- Leslie RGQ: Critical events in the irreversible uptake of soluble immune complexes by macrophages. Immunol Lett 11:153, 1985
-
Wener MH, Mannik M: Mechanisms of immune deposit formation
in renal glomeruli. Springer Semin Immunopathol 9:219, 1986 [PMID
3544280]
-
Van den Dobbelsteen ME, van der Woude FJ, Schroeijers WEM,
et al: Both IgG- and C1q-receptors play a role in the enhanced binding
of IgG complexes to human mesangial cells. J Am Soc Nephrol 7:573, 1996
[PMID
8724891]
-
Wilson JG, Wong WW, Schur PH, et al: Mode of inheritance
of decreased C3b receptors on erythrocytes of patients with systemic lupus
erythematosus. N Engl J Med 307:981, 1982
-
Witte T, Dumoulin F-L, Gessner JE, et al: Defect of a complement
receptor 3 epitope in a patient with systemic lupus erythematosus. J Clin
Invest 92:1181, 1993 [PMID
7690773]
-
Frank MM, Lawley TJ, Hamburger MI, et al: Immunoglobulin
G Fc receptor-mediated clearance in autoimmune diseases. NIH Conference.
Ann Intern Med 98:206, 1983
-
Ruiz P, Gomez F, Schreiber AD: Impaired function of macrophage
Fc-g receptors in end-stage renal disease. N
Engl J Med 322:717, 1990 [PMID
2308600]
-
Steil AA, Garcia Rodriguez MC, Alonso A, et al: Platelet-activating
factor: the effector of protein-rich plasma extravasation and nitric oxide
synthase induction in rat immune complex peritonitis. Br J Pharmacol 114:895,
1995 [PMID
7539698]
-
Sylvestre DL, Ravetch JV: Fc receptors initiate the Arthus
reaction: redefining the inflammatory cascade. Science 265:1095, 1994 [PMID
8066448]
-
ulligan MS, Wilson GP, Todd RF, et al: Role of b1,
b2 integrins and ICAM-1 in lung injury
after deposition of IgG and IgA immune complexes. J Immunol 150:2407, 1993
[PMID
7680691]
-
Shanley TP, Peters JL, Jones ML, et al: Regulatory effects
of endogenous interleukin-1 receptor antagonist protein in immunoglobulin
G immune complex-mediated lung injury. J Clin Invest 97:963, 1996 [PMID
8613550]
-
Heyman B: The immune complex: possible ways of regulating
the antibody response. Immunol Today 11:310, 1990
-
Hornick DB, Fick RB Jr: The immunoglobulin G subclass composition
of immune complexes in cystic fibrosis: implications for the pathogenesis
of the Pseudomonas lung lesion. J Clin Invest 86:1285, 1990 [PMID
2120286]
-
Pier GB, Takeda S, Grout M, et al: Immune complexes from
immunized mice and infected cystic fibrosis patients mediate murine and
human T cell killing of hybridomas producing protective, opsonizing antibody
to Pseudomonas aeruginosa. J Clin Invest 91:1079, 1993
-
Selvaggi TA, Walker RE, Fleisher TA: Development of antibodies
to fetal calf serum with Arthus-like reactions in human immunodeficiency
virus-infected patients given syngeneic lymphocyte infusions. Blood 89:776,
1997 [PMID
9028307]
-
Creamer JD, McGrath JA, Webb-Peploe M, et al: Serum sickness-like
illness following streptokinase therapy: a case report. Clin Exp Dermatol
20:468, 1995 [PMID
8857338]
-
Lawley TJ, Bielory L, Gascon P, et al: A prospective clinical
and immunologic analysis of patients with serum sickness. N Engl J Med
311:1407, 1984 [PMID
6387492]
-
Nityanand S, Holm G, Lefvert AK: Immune complex mediated
vasculitis in hepatitis B and C infections and the effect of antiviral
therapy. Clin Immunol Immunopathol 82:250, 1997
-
Bourgoignie JJ, Pardo V: HIV-associated nephropathies. N
Engl J Med 327:729, 1992 [PMID
1495528]
-
Meryhew NL, Kimberly RP, Messner RP, et al: Mononuclear phagocyte
system in SLE: II. A kinetic model of immune complex handling in systemic
lupus erythematosus. J Immunol 137:97, 1986
-
Davies KA, Peters AM, Beynon HLC, et al: Immune complex processing
in patients with systemic lupus erythematosus. J Clin Invest 90:2075, 1992
[PMID
1430231]
-
Waxman FJ, Hebert LA, Cosio FG, et al: Differential binding
of immunoglobulin A and immunoglobulin G1 immune complexes to primate erythrocytes
in vivo: immunoglobulin A immune complexes bind less well to erythrocytes
and are preferentially deposited in glomeruli. J Clin Invest 77:82, 1986
[PMID
3944261]
-
Greisman SG, Redecha PB, Kimberly RP, et al: Differences
among immune complexes: association of C1q in SLE immune complexes with
renal disease. J Immunol 138:739, 1987 [PMID
3492531]
-
Morioka T, Woitas R, Fujigaki Y, et al: Histone mediates
glomerular deposition of small size DNA anti-DNA complex. Kidney Int 45:991,
1994
-
Suzuki H, Takemura H, Yoshizaki K, et al: IL-6-anti-IL-6
autoantibody complexes with IL-6 activity in sera from some patients with
systemic sclerosis. J Immunol 152:935, 1994 [PMID
8283060]
-
De Benedetti F, Massa M, Pignatti P, et al: Serum soluble
interleukin 6 (IL-6) receptor and IL-6/soluble IL-6 receptor complex in
systemic juvenile rheumatoid arthritis. J Clin Invest 93:2114, 1993
-
Brinkman K, Termaat R, Berden JH, et al: Anti-DNA antibodies
and lupus nephritis: the complexity of crossreactivity. Immunol Today 11:232,
1990 [PMID
2143657]
-
Dersimonian H, Schwartz RS, Barrett KJ, et al: Relationship
of human variable region heavy chain germ-line genes to genes encoding
anti-DNA autoantibodies. J Immunol 139:2496, 1987
-
Barnett LA, Fujinami RS: Molecular mimicry: a mechanism for
autoimmune injury. FASEB J 6:840, 1992 [PMID
1740233]
-
Inman RD, Chiu B, Hamilton NC: Analysis of immune complexes
in rheumatoid arthritis for Epstein-Barr virus antigens reveals cross-reactivity
of viral capsid antigen and human IgG. J Immunol 138:407, 1987 [PMID
3025298]
-
Query CC, Keene JD: A human autoimmune protein associated
with U1 RNA contains a region of homology that is cross-reactive with retroviral
p30gag antigen. Cell 51:211, 1987 [PMID
2959371]
-
Minota S, Koyasu S, Yahara I, et al: Autoantibodies to the
heat-shock protein hsp90 in systemic lupus erythematosus. J Clin Invest
81:106, 1988 [PMID
3335628]
-
Rook GAW, Stanford JL: Slow bacterial infection or autoimmunity?
Immunol Today 13:160, 1992
-
Bartoloni C, Guidi L, Pili R, et al: Assay, isolation and
characterization of circulating immune complexes from serum of gastrointestinal
cancer, stage III and IV melanoma and chronic inflammatory bowel disease
patients. Oncology 50:27, 1993 [PMID
8421596]
-
Waxman FJ, Hebert LA, Cosio FG, et al: Differential binding
of immunoglobulin A and immunoglobulin G1 immune complexes to primate erythrocytes
in vivo. J Clin Invest 77:82, 1985
-
Williams DG: Pathogenesis of idiopathic IgA nephropathy.
Pediatr Nephrol 7:303, 1993
-
Kissel JT, Mendell JR, Rammohan KW: Microvascular deposition
of complement membrane attack complex in dermatomyositis. N Engl J Med
314:329, 1986 [PMID
3945256]
-
Lewis EJ, Hunsicker LG, Lan SP, et al: A controlled trial
of plasmapheresis therapy in severe lupus nephritis. The Lupus Nephritis
Collaborative Study Group. N Engl J Med 326:1373, 1992
-
Miller FW, Leitman SF, Cronin ME, et al: Controlled trial
of plasma exchange and leukapheresis in polymyositis and dermatomyositis.
N Engl J Med 326:1380, 1992 [PMID
1472183]
-
Basta M: Modulation of complement-mediated immune damage
by intravenous immune globulin. Clin Exp Immunol 104(suppl 1):21, 1996
-
Mulligan MS, Warren JS, Smith CW, et al: Lung injury after
deposition of IgA immune complexes: requirement for CD18 and L-arginine.
J Immunol 148:3086, 1992
-
Mulligan MS, Jones ML, Vaporciyan AA, et al: Protective effects
of IL-4 and IL-10 against immune complex-induced lung injury. J Immunol
151:5666, 1993 [PMID
7901280]
-
Mulligan MS, Polley MJ, Bayer RJ, et al: Neutrophil-dependent
acute lung injury: requirement for P-selectin (GMP-140). J Clin Invest
90:1600, 1992 [PMID
1383277]
-
Doherty JB, Ashe BM, Argenbright LW, et al: Cephalosporin
antibiotics can be modified to inhibit leukocyte elastase. Nature 322:192,
1986 [PMID
3636599]
-
Fletcher DS, Osinga DG, Keenan K, et al: An inhibitor of
leukocyte elastase prevents immune complex-mediated hemorrhage in the rat
lung. J Pharmacol Exp Ther 274:548, 1995
-
Kelton JG: Impaired reticuloendothelial function in patients
treated with methyldopa. N Engl J Med 313:595, 1985