Contenido del artículo
II DIAGNOSTICO Y TRATAMIENTO DE LOS TRASTORNOS DE LOS LIPIDOS
- Composición y metabolismo de las lipoproteínas
- Importancia clínica de las alteraciones de los lípidos
- LIPOPROTEINAS DE BAJA DENSIDAD Y ATEROGENESIS
- LBD OXIDADA Y LBD DENSA, PEQUEÑA
- LIPOPROTEINAS DE ALTA DENSIDAD Y ATEROGENESIS
- HIPERTRIGLICERIDEMIA Y ATEROGENESIS
- LP (a) Y ATEROGENESIS
- PANCREATITIS
- Padecimientos primarios del metabolismo de las lipoproteínas
- DEFICIENCIA FAMILIAR DE LIPASA DE LIPOPROTEINA
- DEFICIENCIA FAMILIAR DE APOLIPOPROTEINA C-II
- HIPERTRIGLICERIDEMIA FAMILIAR
- HIPERLIPIDEMIA COMBINADA FAMILIAR
- HIPERCOLESTEROLEMIA FAMILIAR
- DEFECTO FAMILIAR EN LA APOLIPOPROTEINA B-100
- HIPOBETALIPOPROTEINEMIA Y ABETALIPOPROTEINEMIA FAMILIARES
- DISBETALIPOPROTEINEMIA
- SINDROMES PRIMARIOS DE BAJA CONCENTRACION DE LAD
- HIPERCOLESTEROLEMIA POLIGENICA
- Alteraciones secundarias del metabolismo de los lípidos
- Enfoque práctico para los trastornos comunes
- PACIENTES CON AUMENTO DEL COLESTEROL
- PACIENTES CON HIPERLIPIDEMIA COMBINADA
- PACIENTES CON AUMENTO EN LOS TRIGLICERIDOS
- PACIENTES CON BAJA CONCENTRACION DE COLESTEROL DE LAD
- PACIENTES CON ENFERMEDAD CORONARIA Y LIPOPROTEINAS NORMALES
- Tratamiento farmacológico de los trastornos de las lipoproteínas
- FARMACOS PARA EL AUMENTO EN LA CONCENTRACION DE COLESTEROL DE LBD
- FARMACOS USADOS PRINCIPALMENTE PARA EL AUMENTO EN LOS TRIGLICERIDOS
- REMPLAZO ESTROGENICO EN MUJERES POSMENOPAUSICAS
- ACIDOS GRASOS OMEGA-3 (ACEITE DE PESCADO)
- Aspectos especiales del tratamiento de las alteraciones de los lípidos
DR. STEPHEN P. FORTMANN
DR. DAVID J. MARON
Las alteraciones del metabolismo de las lipoproteínas y la susceptibilidad del metabolismo humano a los efectos adversos de las dietas altas en grasas saturadas, de la obesidad y de la falta de actividad física han hecho que la enfermedad ateroesclerótica tenga un carácter epidémico en los Estados Unidos y en otros países desarrollados. A pesar de la disminución en la mortalidad por enfermedad coronaria en los Estados Unidos desde 1970, este problema sigue siendo la primera causa de muerte para ambos sexos.
En forma tradicional, la hiperlipoproteinemia se definía como la elevación de una lipoproteína por arriba de la percentila 95 en la población general. El sistema de clasificación descrito por Fredrickson y colaboradores1 ha sido muy utilizado para describir los fenotipos de hiperlipoproteinemia, pero con frecuencia se utiliza en forma errónea como un esquema de clasificación para los padecimientos primarios, o genéticos, del metabolismo de las lipoproteínas y está cayendo en desuso al conocerse mejor los trastornos primarios. El reconocimiento de que un nivel bajo de lipoproteína de alta densidad (LAD) tiene importancia clínica ha popularizado el término de dislipoproteinemia (que parece ser el más adecuado) para describir un grupo de padecimientos con lipoproteínas demasiado altas o bajas.
Las dislipoproteinemias tienen importancia clínica por su contribución a la aterogénesis. El riesgo de enfermedad vascular ateroesclerótica comienza a aumentar con concentraciones de colesterol de alrededor de 180 mg/dl,2 un nivel muy por debajo de la percentila 95 que se utilizaba antes para definir la hipercolesterolemia. Alrededor de la mitad de la población en los Estados Unidos tiene una concentración de colesterol que aumenta su riesgo en forma significativa. Por ello, el National Cholesterol Education Program (NCEP) (Programa nacional de educación sobre el colesterol, n. del t.) ha modificado la definición de hipercolesterolemia a cualquier nivel superior a 200 mg/dl. Desde el punto de vista de los autores, el nivel óptimo de colesterol debe ser menor de 150 mg/dl.
Composición y metabolismo de las lipoproteínas
Las lipoproteínas son complejos esféricos macromoleculares de lípidos y proteína [ver figura 1]. Los constituyentes lípidos de las lipoproteínas son colesterol esterificado y no esterificado (libre), triglicéridos y fosfolípidos. El componente proteico se denomina apolipoproteína. Las lipoproteínas transportan colesterol y triglicéridos (que no son hidrosolubles) desde los sitios de absorción y síntesis hasta los sitios de utilización. El éster colesteril y los triglicéridos son no polares y constituyen el centro hidrofóbico de las lipoproteínas en proporciones variables. La superficie de la lipoproteína contiene moléculas polares (colesterol libre, fosfolípidos y apolipoproteínas), lo que permite que estas partículas se combinen con el plasma mientras transportan su cargamento hidrofóbico. Las lipoproteínas se clasifican en cinco grupos principales de acuerdo con su tamaño, densidad, movimiento electroforético y composición de lípidos y proteínas [ver tabla 1]. Las apolipoproteínas proporcionan estabilidad estructural, actúan como cofactores para enzimas específicas o como ligandos para receptores específicos relacionados con el metabolismo de las lipoproteínas [ver tabla 2].
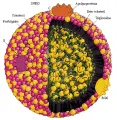
|
| Figura 1 |
| Lipoproteína de muy baja densidad |
|
||||||||||||
|
|
||||||||||||||||||||||
|
El colesterol se utiliza para la síntesis de ácidos biliares en el hígado, la fabricación y reparación de membranas celulares y para la síntesis de las hormonas esteroides. Las fuentes de colesterol son tanto exógenas (dieta) como endógenas (hígado principalmente). El individuo norteamericano promedio consume alrededor de 300 mg de colesterol cada día, y produce 500 a 1,000 mg adicionales en el hígado y otros tejidos. Otra fuente de colesterol son los 500 a 1,000 mg de colesterol biliar que se secretan en el intestino cada día, de los cuales se reabsorbe el 50 porciento (circulación enterohepática). Los triglicéridos, que son lípidos no polares formados por un esqueleto glicerol y tres ácidos grasos de longitud y grado de saturación variables, se utilizan como reserva en el tejido adiposo y para la obtención de energía. Se localizan en el hígado y los adipocitos, y también derivan de fuentes dietéticas y del hígado.
El sistema de transporte de lípidos se divide en la vía exógena, para los triglicéridos y colesterol absorbidos en el intestino, y la vía endógena, para los triglicéridos y colesterol secretados por el hígado [ver figura 2]. El sistema de transporte inverso de colesterol, mediado por LAD, participa en ambas vías. Tres enzimas principales, la lipasa de lipoproteína, la lecitín-colesterol aciltransferasa (LCAT) y la lipasa de triglicéridos hepáticos, participan en el metabolismo de las lipoproteínas [ver tabla 3].
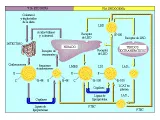
|
| Figura 2 |
| Vías de transporte de los lípidos |
|
||||||||||||
|
VIA EXOGENA
Después de una comida, las células intestinales absorben los ácidos grasos y el colesterol, los esterifican para formar triglicéridos y éster colesteril, y los incorporan a un núcleo de quilomicrones [ver figura 2]. Los triglicéridos predominan, con mucho, sobre los ésteres colesteril en el núcleo. Los quilomicrones penetran a la circulación venosa por los vasos linfáticos. La apolipoproteína C-II (apo C-II) activa a la lipasa de lipoproteína endotelial, que hidroliza los triglicéridos del núcleo del quilomicrón. Los ácidos grasos libres resultantes son captados por el tejido adiposo para su almacenamiento y por el músculo para obtener energía. Durante la lipólisis el quilomicrón se encoge, además los componentes de su superficie protruyen y son transferidos a las LAD en intercambio por apolipoproteínas adicionales y ésteres colesteril. En condiciones normales, los quilomicrones están presentes en el plasma sólo algunos minutos.3 Los quilomicrones residuales tienen mayor cantidad relativa de ésteres colesteril, tanto por haber perdido triglicéridos como por tener ésteres adicionales provenientes de las LAD. Estos residuos son eliminados con rapidez del plasma por dos receptores en el hígado que se unen en forma ávida a la apo E: el receptor de la lipoproteína de baja densidad (LBD) y un receptor aparentemente único de residuos de quilomicrón denominado proteína relacionada al receptor de LBD, o PRB. Una vez dentro del hepatocito, las partículas residuales son digeridas por enzimas lisosomales y el colesterol es esterificado y almacenado. A continuación, el éster colesteril intracelular es usado para la síntesis de ácidos biliares, de membranas celulares o de lipoproteínas por la vía endógena. También se excreta colesterol libre a través de la bilis.
VIA ENDOGENA
Las lipoproteínas de muy baja densidad (LMBD) son lipoproteínas de gran tamaño, ricas en triglicéridos, que se sintetizan y secretan en los hepatocitos [ver figura 2]. El triglicérido de las LMBD es elaborado a partir de glicerol y ácidos grasos que han sido liberados por el tejido adiposo o sintetizados por el hígado. El colesterol de las LMBD proviene de las lipoproteínas circulantes o de la síntesis hepática. Al igual que en el caso de los quilomicrones, las LMBD interactúan con la lipasa de lipoproteína en el endotelio capilar, y los triglicéridos centrales son hidrolizados para proporcionar ácidos grasos a los tejidos adiposo y muscular. Alrededor de la mitad de las partículas catabolizadas de LMBD (en ocasiones llamadas remanentes de LMBD) son captadas por los receptores hepáticos de LBD, y la otra mitad permanece en el plasma, convirtiéndose en lipoproteínas de densidad intermedia (LDI). Las LDI retienen las apoproteínas B-100 y E, tienen mayor cantidad relativa de ésteres colesteril, menor cantidad de triglicéridos, y circulan de minutos a horas, siendo convertidas en forma gradual por la lipasa hepática de triglicéridos, a LBD, más pequeñas, más densas y ricas en ésteres colesteril. Al convertirse las LDI en LBD, la apolipoproteína E se libera y sólo queda una apolipoproteína en la partícula, la apo B-100.
En condiciones normales las LBD transportan alrededor del 75 porciento del colesterol circulante, llevándolo a las células extrahepáticas para la síntesis de hormonas esteroides y membranas celulares. Las partículas de LBD circulan durante aproximadamente dos a tres días,3 y la mitad de éstas son captadas por los receptores de LBD del hígado. La captación de LBD es menor que la de lipoproteínas que contienen apo E porque el receptor de LBD tiene menor afinidad para la apo B-100 que para la apo E. La captación celular de las LBD está mediada por una molécula receptora glucoproteica que fija la apo B-100.4 Alrededor del 70 porciento de las LBD son depuradas por captación por el receptor, y el resto son eliminadas por un mecanismo destructor de la célula. Después de que la LBD es captada por receptores, se libera colesterol y se acumula dentro de las células, con tres consecuencias metabólicas importantes.4 Primero, disminuye la síntesis de 3-hidroxi- 3-metilglutaril coenzima A (HMG-CoA) reductasa, que es la enzima que controla la velocidad de la síntesis de colesterol de novo dentro de la célula. Segundo, se activa la enzima acil colesterol aciltransferasa (ACAT), que esterifica el colesterol libre para formar ésteres de colesteril, que es la forma en que se almacena éste dentro de la célula. Tercero, el acúmulo de colesterol suprime la síntesis celular de nuevos receptores para LBD. Este mecanismo de retroalimentación reduce la captación celular de LBD de la circulación [ver figura 3].
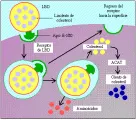
|
| Figura 3 |
| Receptor de lipoproteínas de baja densidad |
ORIGEN Y METABOLISMO DE LA LAD
El origen y metabolismo de la LAD no se conoce tan bien como el de otras lipoproteínas, y parece ser más complejo y variado.5 Las principales apolipoproteínas en la LAD son apo A-I y apo A-II, que se elaboran en el hígado y el intestino delgado. Estas apolipoproteínas son secretadas con fosfolípidos en una estructura parecida a un disco llamada LAD naciente. La mayoría de las apolipoproteínas y fosfolípidos destinados a convertirse en LAD naciente se secretan al inicio en la superficie de los quilomicrones y la LMBD y se transfieren a la LAD durante la lipólisis. Al asociarse las LAD pobres en lípidos con células, atraen colesterol de las membranas celulares y el colesterol difunde al interior de la cubierta de superficie de la LAD. La enzima circulante LCAT se asocia con la LAD y es activada por la apo A-I para esterificar colesterol libre en la superficie de la LAD, causando que se mueva hacia el núcleo. Estas partículas se vuelven esféricas y se denominan LAD3. Cuando las partículas de LAD3 acumulan ésteres colesteril en el núcleo y componentes de la superficie de los quilomicrones y LMBD, se convierten en partículas LAD2 enriquecidas con ésteres colesteril. La mayor parte de los ésteres colesteril de las partículas LAD2 son entonces transferidas a lipoproteínas ricas en triglicéridos intercambiándose por triglicéridos, en un proceso mediado por una proteína de transferencia de ésteres colesteril.6 Los triglicéridos adquiridos por la LAD pueden ser hidrolizados por acción de la lipasa hepática de triglicéridos, convirtiendo a la LAD2 en LAD3, que vuelve a estar lista para aceptar más colesterol libre de las membranas celulares. Las LAD2 ricas en ésteres colesteril y con apo E pueden también ser captadas en forma directa por receptores hepáticos, pero el mecanismo por el que esto sucede no es claro. También es posible que la LAD2 proporcione ésteres colesteril en forma directa al hígado sin que exista catabolismo de la partícula de LAD. A todo este proceso se le denomina transporte inverso del colesterol.
Se piensa que la LAD es antiaterogénica por el transporte inverso de colesterol. Las partículas de LAD pueden dividirse en dos categorías: las que contienen solo apo A-I y las que contienen tanto apo A-I como apo-A-II. La salida de colesterol de los tejidos extrahepáticos parece ser mediada principalmente por partículas de LAD que contienen solo apo A-I.7 La expresión exagerada de apo A-I humana en ratones transgénicos inhibe la ateroesclerosis,8 y la expresión exagerada de apo A-II la promueve.9 También se ha observado que la LAD inhibe la oxidación de LBD, otro mecanismo potencial por el que la LAD puede disminuir la ateroesclerosis.10
LIPOPROTEINA(a)
La lipoproteína(a) [Lp(a)] es una clase específica de partícula de lipoproteína sintetizada en el hígado y con composición de lípidos muy semejante a la de la LBD. La Lp(a) difiere de la LBD por la presencia de una proteína altamente glucosilada de masa variable denominada apo(a), unida por un enlace covalente a la apo B-100. La investigación sobre la Lp(a) y la ateroesclerosis se aceleró al descubrir la homología estructural entre la apo(a) y el plasminógeno, una proteína clave en la cascada de la coagulación. Se sabe poco sobre el catabolismo de la Lp(a) o su papel fisiológico.
Las concentraciones plasmáticas de Lp(a) varían mucho entre los individuos, desde niveles casi no detectables hasta 200 mg/dl. La concentración de Lp(a) parece estar controlada en forma estrecha por factores genéticos.11
Importancia clínica de las alteraciones de los lípidos
Las principales manifestaciones clínicas de los padecimientos comunes de los lípidos son enfermedad cardiovascular ateroesclerótica y pancreatitis. Las lipoproteínas participan en forma fundamental en la aterogénesis y la enfermedad vascular subsecuente es la causa más importante de muerte e incapacidad en los Estados Unidos. La pancreatitis es una manifestación mucho menos común pero clínicamente severa de los niveles de triglicéridos.
LIPOPROTEINAS DE BAJA DENSIDAD Y ATEROGENESIS
Estudios epidemiológicos, de autopsia y en animales han establecido con firmeza que la concentración elevada de LBD es aterogénica. El receptor de la LBD se une a la LBD en forma óptima cuando la concentración de colesterol en el líquido intersticial es de 2.5 mg/dl, que corresponde a una concentración de colesterol de LBD en plasma de 25 mg/dl4 [ver figura 4]. Los recién nacidos tienen una concentración de colesterol de LBD de alrededor de 30 mg/dl, y los mamíferos que no desarrollan ateroesclerosis y las personas con dietas muy bajas en grasas, semejantes a las de nuestros antecesores,12 tienen concentraciónes de colesterol de LBD en plasma menores de 80 mg/dl. El adulto varón promedio en los Estados Unidos tiene una concentración de colesterol en plasma de 125 mg/dl, lo que proporciona un amplio suplemento para la aterogénesis. Esta susceptibilidad humana general a las grasas saturadas de la dieta causa hipercolesterolemia y enfermedad arterial coronaria epidémica.13 El abordaje adecuado para controlar la ateroesclerosis epidémica debe ser a través de la salud pública, por medios como educación, políticas agrícolas y etiquetación de alimentos.14 El papel de la comunidad médica en este esfuerzo debe ser insistir en los mensajes sobre dieta y ejercicio, e identificar a las personas con riesgo suficiente para justificar una intervención especial. Los riesgos del tratamiento deben balancearse con la posibilidad de beneficio y el tratamiento debe ser individualizado. La mayoría de las personas sin enfermedad vascular conocida pueden tratarse con una dieta más saludable.

|
| Figura 4 |
| Niveles de lipoproteínas de baja densidad |
Los trabajos de prevención primaria usando terapia farmacológica han demostrado que los eventos coronarios se reducen en forma significativa disminuyendo la concentración de LBD en varones hiperlipidémicos asintomáticos.15-17 El estudio más reciente y extenso al respecto fue el primero que empleó un inhibidor potente de la HMG-CoA reductasa. Incluyó 6,595 varones de 45 a 64 años de edad con una concentración promedio basal de LBD de 192 mg/dl que recibieron pravastatina, 40 mg/día, o placebo.16 Después de un promedio de 4.9 años, el grupo con tratamiento activo presentó una reducción del 31 porciento en la incidencia de muerte coronaria o infarto al miocardio (IM) no fatal (riesgo absoluto de 5.5 contra 7.9 porciento), una reducción del 32 porciento en muertes cardiovasculares, una reducción del 38 porciento en procedimientos cardiovasculares y una reducción del 22 porciento en la mortalidad por todas las causas. Además, varios estudios angiográficos aleatorios han comparado los efectos del tratamiento agresivo de los lípidos con los del placebo o el tratamiento habitual sobre la progresión de la ateroesclerosis coronaria tanto en hombres como en mujeres.18 Aunque estos estudios varían mucho en la selección y tamaño de muestra de los pacientes, tipo de tratamiento, duración de la intervención y puntos finales angiográficos, se han encontrado resultados coherentes. La reducción significativa en la LBD, en el rango de 25 a 40 porciento, se asocia con progresión significativamente menor, mayor regresión de la ateroesclerosis en la angiografía de seguimiento y menos eventos cardiacos clínicos.
El Scandinavian Simvastatin Survival Study (4S)19 (Estudio escandinavo de supervivencia con simvastatin, n. del t.), incluyó 4,444 pacientes (81 porciento hombres) con hipercolesterolemia (LBD basal promedio de 188 mg/dl) e historia de infarto al miocardio o angina, que recibieron simvastatina o placebo. Después de 5.4 años de seguimiento, el grupo tratado experimentó una reducción del 30 porciento en la mortalidad total (ocho porciento en el grupo de simvastatina contra 12 porciento en el grupo placebo), una reducción del 42 porciento en la mortalidd coronaria, 37 porciento menos necesidad de cirugía de derivación coronaria y angioplastía transluminal percutánea y 30 porciento menos eventos cerebrovasculares. El Cholesterol and Recurrent Events (CARE) trial20 (Estudio sobre colesterol y eventos recurrentes, n. del t.) distribuyó 4,159 pacientes (86 porciento varones) con hipercolesterolemia moderada (LBD basal promedio de 139 mg/dl) e historia de infarto del miocardio para recibir pravastatina o placebo. Después de cinco años el grupo tratado con pravastatina tuvo una reducción del 24 porciento en los eventos coronarios fatales y no fatales (10.2 porciento contra 13.2 porciento). El grupo tratado tuvo también una reducción del 27 porciento en la necesidad de procedimientos de revascularización coronaria y un 31 porciento de reducción en la incidencia de eventos cerebrovasculares.
Recientemente se ha revisado la patogenia de los síndromes coronarios agudos y los posibles mecanismos por los que la reducción de la LBD puede producir beneficios clínicos.21,22 La fisura y ruptura de placas ateroescleróticas ricas en lípidos causa agregación plaquetaria, vasoespasmo y trombosis intraluminal. Muchos estudios angiográficos han reportado significativamente menos eventos cardiacos clínicos aún con beneficios angiográficos mínimos (aunque mesurables) en los pacientes que reciben tratamiento intensivo para el control de los lípidos. Este dato es compatible con el concepto de la estabilización de placas, que consiste en que el control intensivo de los lípidos depleta éstos del núcleo de las placas, reduce la concentración de LBD oxidada y células inflamatorias, fortelece la fuerza tensil de la capa fibrosa y hace que las placas se fracturen menos.18 Es posible que la estabilización de las placas, más que la regresión geométrica de las mismas o el aumento en la luz, sea el objetivo más imporante de la disminución agresiva de los niveles de LBD.
El endotelio vascular es responsable de la liberación de factor de relajación derivado del endotelio (FRDE), identificado como óxido nítrico (NO) y prostaciclina (PGI2). El FRDE-NO y la PGI2 inhiben la contracción del músculo liso, la proliferación de las células musculares lisas, la agregación plaquetaria y la adhesión de plaquetas y monocitos a la superficie endotelial.23 La función endotelial se deteriora por la hipercolesterolemia24 y mejora al disminuir el colesterol.25,26 Debido a que la agregación plaquetaria y el vasoespasmo coronario tienen funciones importantes en los síndromes coronarios agudos, la reducción del colesterol puede disminuir la incidencia de eventos cardiacos agudos al restablecer la función endotelial. El tratamiento de los pacientes con enfermedad arterial coronaria con estatinas reduce la frecuencia y duración de los episodios de isquemia medidos por vigilancia ECG ambulatoria.27,28
Esta importante convergencia de datos epidemiológicos, de ciencia básica y clínicos, con los resultados de los estudios clínicos a gran escala ha llevado a un claro consenso científico respecto a que el tratamiento de los trastornos de los lípidos es la piedra angular en la prevención primaria y secundaria de las enfermedades vasculares.29
LBD OXIDADA Y LBD DENSA, PEQUEÑA
Además de la concentración de LBD, otros dos aspectos sobre la LBD han sido objeto de investigación importante: la oxidación de la LBD y sus subclases. Se ha encontrdo que la LBD debe sufrir modificaciones antes de ser ingerida por los macrófagos para formar células espumosas, que son componentes importantes de las placas ateroescleróticas.30 In vivo, la modificación oxidativa es quizá la forma más frecuente de modificación. La LBD oxidada no solo promueve la formación de células espumosas, sino que también es quimiotáctica para los monocitos circulantes, es citotóxica y altera la función endotelial. Se ha demostrado que el tratamiento antioxidante mejora la función de las células endoteliales en pacientes con hipercolesterolemia y enfermedad arterial coronaria.26 El Cambridge Heart Antioxidant Study (CHAOS, Estudio sobre antioxidantes cardiacos de Cambridge, n. del t.) distribuyó 2,002 pacientes con enfermedad coronaria demostrada por angiografía para recibir vitamina E, 400 a 800 UI o placebo. Después de un seguimiento promedio de 1.4 años, el tratamiento antioxidante redujo el punto final primario de muerte cardiovascular e IM no fatal en 47 porciento (41 eventos contra 64).31 En la actualidad se realizan estudios clínicos extensos sobre el efecto de los antioxidantes en los eventos cardiovasculares adversos.
Es posible distinguir en la población dos patrones de subclases diferentes de LBD, denominadas A y B, esto se realiza por ultracentrifugación y gradiente de electroforesis en gel.32 Las personas con el patrón A tienen principalmente partículas de LBD grandes, enriquecidas con éster colestril, mientras que las que tienen patrón B tienen partículas de LBD densas y pequeñas, enriquecidas con triglicéridos. Las LBD densas y pequeñas tienen más propensión a oxidarse. El patrón B se asocia con niveles mayores de triglicéridos y apo B, menor cantidad de LAD y apo A-I y mayor riesgo de enfermedad arterial coronaria. Este patrón B parece heredarse como un rasgo unigénico y se asocia en forma estrecha con hiperlipidemia combinada familiar.33 La relación colesterol total:LAD y la concentración de triglicéridos han demostrado en estudios prospectivos ser predictores más intensos de enfermedad arterial coronaria que el tamaño de la LBD; por lo tanto, los autores no recomiendan medir de rutina el tamaño de las LBD.34
LIPOPROTEINAS DE ALTA DENSIDAD Y ATEROGENESIS
Algunos síndromes hereditarios de LAD bajas se asocian con enfermedad coronaria prematura, pero otros no [ver adelante, Síndromes primarios de LAD baja]. Los estudios epidemiológicos han demostrado que, dentro de un grupo de población, la concentración de LAD correlaciona en forma estrecha e inversa con el riesgo de enfermedad coronaria, independientemente del nivel de colesterol de LBD. Tanto la concentración de LAD2 como la de LAD3 se relacionan en forma inversa con el riesgo de enfermedad cardiaca.35 El mecanismo para este efecto protector potencial de la LAD se desconoce, pero puede deberse al transporte inverso del colesterol y a la capacidad de la LAD para inhibir la oxidación de la LBD.10 La importancia de la LAD cuando la concentración de LBD es muy baja no es clara; es posible que la LAD tenga menos importancia en estos casos porque cuando la concentración de LBD en la circulación es baja, el papel de las LAD en el transporte inverso del colesterol es menos crucial. Cuando se comparan los niveles de LAD entre las poblaciones, los países con concentraciones promedio más altas suelen tener mayores porcentajes de enfermedad coronaria que las naciones con concentraciones promedio menores. Esto parece deberse a que la LAD y la LBD tienen una correlación directa en los grupos de población, y ambas se relacionan en forma positiva con la ingesta de grasa en la dieta.36 En los conejos alimentados con colesterol, la administración de LAD homóloga inhibe la formación de estrías grasas aórticas y reduce la ateroesclerosis aórtica establecida.37,38
Dos estudios de prevención primaria15,17 encontraron un beneficio aparente al aumentar la concentración de LAD que fue independiente de la disminución de la LBD, pero ambos estudios emplearon fármacos que disminuyen también la LBD, y ninguno incluyó individuos seleccionados por tener LAD baja. Un estudio con control de casos realizado en varones con enfermedad coronaria demostrada por angiografía encontró que más de una tercera parte de los pacientes tenía concentración de colesterol total por debajo de 200 mg/dl, y de estos, casi tres cuartas partes tenían concentraciones de LAD menores de 35 mg/dl.39 La vigilancia de estos pacientes demostró que tienen un gran riesgo de sufrir eventos cardiovasculares subsecuentes.40 Los estudios que han usado angiografía coronaria para demostrar la progresión de la ateroesclerosis en hombres con enfermedad coronaria han demostrado que el uso de intervenciones farmacológicas para aumentar la concentración de LAD evita la progresión o induce regresión de la ateroesclerosis.41,42 Las normas de la American Heart Association recomiendan que se considere administrar tratamiento para aumentar los niveles de LAD en los pacientes con enfermedad arterial coronaria que tengan un nivel de LAD bajo. El tratamiento debe comenzar con medidas no farmacológicas, como suspensión del tabaquismo, disminución de peso, y realización de ejercicio. Si se requiere tratamiento farmacológico en estos pacientes para alcanzar una concentración de LBD menor de 100 mg/dl, se recomienda usar niacina o una estatina.43 Tanto el estudio 4S como el CARE encontraron que los individuos con una concentración basal baja de LAD tuvieron beneficios clínicos al recibir tratamiento con estatinas.20,44 El tratamiento farmacológico de los pacientes asintomáticos con LAD baja pero concentraciones normales de LBD es motivo de controversia porque no existen datos de estudios clínicos al respecto.
HIPERTRIGLICERIDEMIA Y ATEROGENESIS
No existen evidencias suficientes para concluir que la hipertrigliceridemia cause enfermedad coronaria. Los lineamientos más recientes del NCEP29 clasifican los triglicéridos como normales (< 200 mg/dl), en límite alto (200 a 400 mg/dl), altos (400 a 1,000 mg/dl) y muy altos (> 1,000 mg /dl). En muchos estudios de población la hipertrigliceridemia se asocia con enfermedad coronaria en los análisis univariados, pero esta relación se debilita o desaparece cuando se consideran los triglicéridos junto con otros factores de riesgo, como concentración de LBD, concentración de LAD, presión arterial, actividad física y obesidad.45 Un estudio observacional mostró que las concentraciones de triglicéridos por arriba de 200 mg/dl aumentaban en forma significativa el riesgo de enfermedad coronaria en los individuos con concentraciones elevadas de LBD y bajas de LAD.46 La ateroesclerosis prematura se asocia con algunos trastornos hereditarios de hipertrigliceridemia (v.gr., hiperlipidemia y disbetalipoproteinemia familiar combinada) pero no con otros (v.gr., deficiencia de lipasa de lipoproteína e hipertrigliceridemia familiar). Las placas ateroescleróticas no contienen triglicéridos, y no se han realizado estudios clínicos diseñados para estudiar el efecto de la disminución de la concentración de triglicéridos sobre la enfermedad coronaria. Por lo general, los estudios clínicos y angiográficos en los que se disminuye el colesterol total o el colesterol de LBD, han encontrado que la disminución de los triglicéridos no tuvo un efecto significativo sobre la evolución clínica o angiográfica. Sin embargo, el Helsinky Heart Study (Estudio del corazón de Helsinky, n. del t. )que se dedicó sobre todo al aspecto de prevención, encontró que los individuos con concentración elevada de LBD, baja concentración de LAD y nivel de triglicéridos por arriba de 200 mg/dl, tuvieron los mayores beneficios por el tratamiento con gemfibrozil, un agente que disminuye el nivel de triglicéridos y aumenta la concentración de LAD.47 Además, el Stockholm Ischaemic Heart Disease Secondary Prevention Study (Estudio de Estocolmo para la prevención secundaria de la enfermedad isquémica del corazón, n. del t.) encontró que la disminución de los niveles de triglicéridos con clofibrato y ácido nicotínico redujo la mortalidad global y coronaria.48
Es probable que la elevación per se de triglicéridos no cause ateroesclerosis, sino que ésta se deba a las alteraciones metabólicas asociadas con la hipertrigliceridemia. Es frecuente que la hipertrigliceridemia se asocie con concentraciones elevadas de quilomicrones, remanentes de LMBD, partículas pequeñas y densas de LBD y disminución en los niveles de LAD, y todos estos trastornos facilitan la aterogénesis.32 Los individuos con baja concentración de LAD y alta de triglicéridos pueden tener un riesgo especialmente alto de enfermedad coronaria.49 Las concentraciones elevadas de triglicéridos también se asocian con aumento de ciertos factores de la coagulación y con menor actividad fibrinolítica. Por último, las concentraciones de triglicéridos en ayuno pueden revelar la eficacia con la que un individuo elimina los triglicéridos posprandiales. Cuando este proceso es ineficaz, la lipemia posprandial prolongada puede inducir captación de remanentes ricos en triglicéridos por las células arteriales.50
LP (a) Y ATEROGENESIS
Algunos estudios epidemiológicos sugieren que la concentración de Lp(a) constituye un factor de riesgo para la enfermedad coronaria y el evento cerebrovascular,51-54 pero otros no muestran relación.55,56 Posibles explicaciones para esta discrepancia aparente incluyen diferencias en los métodos de medición y la posibilidad de que la Lp(a) pueda aumentar el riesgo solo entre las personas con hipercolesterolemia.57 Si la Lp(a) es aterogénica, puede ser por sus propiedades semejantes a LBD: se ha demostrado que la Lp(a) tiene un alto grado de homología con el plasminógeno y puede tener actividad protrombótica al interferir con la unión del plasminógeno a la fibrina. Los datos preliminares sugieren que la reducción del colesterol de LBD elevado en pacientes con niveles altos de Lp(a) puede ser una estrategia eficaz para reducir la progresión de la ateroesclerosis y prevenir eventos coronarios. La Lp(a) en sí puede disminuirse con niacina, estrógenos, tamoxifen o aféresis de LBD. Sin embargo, existen pocos datos sobre la eficacia de disminuir el nivel de Lp(a) para inhibir la ateroesclerosis o prevenir eventos coronarios, y no es posible recomendar el escrutinio o tratamiento de rutina de los niveles elevados de Lp(a).57
PANCREATITIS
Después de la aterogénesis, la pancreatitis aguda es la principal manifestación clínica de la dislipidemia, y se asocia con quilomicronemia y aumento en la concentración de LMBD. La mayoría de los pacientes con pancreatitis tienen concentraciones de triglicéridos superiores a 2,000 mg/dl, pero debe comenzarse tratamiento profiláctico cuando la concentración en ayuno es mayor a 500 mg/dl.58 El mecanismo por el cual la quilomicronemia y el aumento en las LMBD causan pancreatitis es poco claro. Es posible que la lipasa pancreática actúe sobre los triglicéridos dentro de los capilares pancreáticos, causando la formación de ácidos grasos tóxicos que provocan inflamación.
Padecimientos primarios del metabolismo de las lipoproteínas
Los padecimientos primarios o familiares del metabolismo de las lipoproteínas se originan de defectos genéticos en sus vías metabólicas. Los padecimientos principales son la deficiencia familiar de lipasa de lipoproteína, la deficiencia familiar de apolipoproteína C-II, la hipertrigliceridemia familiar, la hiperlipidemia familiar combinada, la hipercolesterolemia familiar, la apolipoproteína B-100 defectuosa familiar, la hipobetalipoproteinemia familiar y los síndromes primarios de abetalipoproteinemia, disbetalipoproteinemia y LAD baja.
DEFICIENCIA FAMILIAR DE LIPASA DE LIPOPROTEINA
La deficiencia familiar de lipasa de lipoproteína es un padecimiento poco frecuente, autosómico recesivo (con un homocigoto por millón de personas), causado por la mutación del gen LPL que provoca una deficiencia severa o ausencia de la lipasa de lipoproteína. En este padecimiento la concentración de apo C-II es normal. Después de una comida con grasa, los quilomicrones se producen en forma normal, pero no son catabolizados, por lo que se acumulan en forma masiva en el plasma, causando hipertrigliceridemia grave que puede persistir durante varios días. La concentración de LMBD suele no estar elevada, y el nivel de LAD y de LBD es bajo. Se ha notificado una variante de esta enfermedad en una familia con un inhibidor circulante de la lipasa de lipoproteína.59
La enfermedad se detecta, en forma típica, durante la niñez temprana por episodios recurrentes de dolor abdominal secundarios a pancreatitis asociada con hipertrigliceridemia (1,000 a 5,000 mg/dl) y quilomicronemia severas. Durante los episodios agudos de pancreatitis la concentración sérica y urinaria de amilasa puede ser normal por interferencia de la hipertrigliceridemia o por inhibidores circulantes de la actividad de amilasa. Los individuos afectados tienen hepatoesplenomegalia por acúmulo de triglicéridos en las células reticuloendoteliales. También pueden existir xantomas eruptivos en los glúteos y superficies extensoras de las extremidades. La quilomicronemia severa se asocia con disnea, aunque no se conoce el motivo de esto.60 El síndrome de quilomicronemia, que consiste en pancreatitis, organomegalia, xantomas eruptivos y lipemia retinalis, es reversible al normalizarse la concentración de triglicéridos. El riesgo de ateroesclerosis puede estar aumentado,61 y la pancreatitis recurrente puede ocasionar insuficiencia pancreática. Los heterocigotos para la deficiencia familiar de lipasa de lipoproteína pueden tener aumento en la concentración de LMBD o LBD, y un síndrome clínico semejante o idéntico a la hiperlipidemia familiar combinada.60,62
Deberá sospecharse este padecimiento en individuos jóvenes con plasma lipémico en ayuno y dolor abdominal grave. El diagnóstico se realiza al demostrar disminución importante o ausencia de la actividad de lipasa de lipoproteína después de la infusión de heparina (que causa la liberación de la lipasa de lipoproteína unida a la pared endotelial) o midiendo la actividad de lipasa de lipoproteína en una biopsia de tejido adiposo. El ensayo debe contener plasma normal o apo C-II. El diagnóstico puede confirmarse demostrando un defecto en el gen LPL.
El tratamiento está orientado a disminuir la formación de quilomicrones por medio de una dieta con restricción absoluta de grasas a menos de 10 a 20 g/día. Pueden proporcionarse calorías grasas adicionales en forma de suplemento de triglicéridos de cadena mediana porque éstos no se incorporan a los quilomicrones, sino que se absorben en forma directa hacia la vena porta. Deberá restringirse la ingesta de alcohol y de estrógenos exógenos. La meta del tratamiento es disminuir la concentración de triglicéridos en ayuno a menos de 1,000 mg/dl. Además del tratamiento dietético, en ocasiones es útil la administración de gemfibrozil o de ácidos grasos omega-3. Durante los episodios de pancreatitis aguda se suspenderá la alimentación por vía oral y el paciente recibirá nutrición intravenosa sin lípidos. En casos de pancreatitis grave durante el embarazo, la plasmaféresis aguda reduce las concentraciones de triglicéridos y los síntomas en forma muy eficaz.63
DEFICIENCIA FAMILIAR DE APOLIPOPROTEINA C-II
La deficiencia familiar de apolipoproteína C-II es un padecimiento autosómico recesivo poco frecuente (un homocigoto por cada millón de personas) causado por una mutación genética que ocasiona deficiencia severa o ausencia de apo C-II. Esta mutación provoca deficiencia funcional de la lipasa de lipoproteína y un síndrome clínico de hiperquilomicronemia. Los pacientes con deficiencia familiar de apo C-II tienden a ser de más edad y tener hipertrigliceridemia menos severa que los pacientes con deficiencia familiar de lipasa de lipoproteína. El diagnóstico se confirma demostrando que el plasma del individuo afectado es incapaz de activar in vitro a la lipasa de lipoproteína o determinando la presencia de deficiencia de apo C-II por medio de enfoque isoeléctrico o electroforesis en gel de dos dimensiones. La actividad de la lipasa de lipoproteína después de la infusión de heparina es normal si se usa apo C-II exógena. Los individuos heterocigotos tienen el 50 porciento de la concentración normal de apo C-II y pueden sufrir hipertrigliceridemia leve, pero no desarrollan quilomicronemia ni pancreatitis. El tratamiento consiste en restringir la grasa de la dieta a menos de 20 g/día. Los pacientes con pancreatitis aguda pueden ser tratados con infusión del plasma normal (que contiene apo C-II) o con apo C-II purificada, que causa un rápido descenso en la concentración de triglicéridos.
HIPERTRIGLICERIDEMIA FAMILIAR
La hipertrigliceridemia familiar es una enfermedad autosómica dominante bastante frecuente, afectando a uno o dos porciento de la población. Existe una mayor síntesis de triglicéridos y las partículas de LMBD están enriquecidas con triglicéridos, aunque no se secretan en un número excesivo.64 Los individuos afectados tienen aumento en la concentración de LMBD, pero niveles bajos de LBD y de LAD, y suelen estar asintomáticos a menos de que desarrollen hipertrigliceridemia grave. La hipertrigliceridemia familiar no parece asociarse con un mayor riesgo de enfermedad coronaria.60 El diagnóstico se realiza por los antecedentes familiares y el perfil de las lipoproteínas en ayuno del paciente y de sus familiares. La concentración de triglicéridos puede variar entre 250 a 1,000 mg/dl en alrededor de la mitad de los parientes en primer grado, puede no existir historia familiar de enfermedad coronaria prematura, ni hipercolesterolemia severa. Si es necesario, los pacientes deberán reducir de peso, realizar ejercicio regular y disminuir su ingesta de ácidos grasos saturados y de colesterol. Se prohibirán el alcohol, los estrógenos exógenos y otros fármacos que aumentan la concentración de LMBD. Si existe diabetes, esta deberá controlarse en forma estricta. La hipertrigliceridemia de este padecimiento suele responder a estas medidas, pero si la concentración de triglicéridos es mayor a 500 mg/dl después de seis meses de tratamiento no farmacológico, deberá iniciarse tratamiento con gemfibrozil o ácido nicotínico. No debe administrarse ácido nicotínico en pacientes diabéticos. Los ácidos grasos omega-3 pueden también ser útiles.
HIPERLIPIDEMIA COMBINADA FAMILIAR
La hiperlipidemia combinada familiar (HCF) fue considerada en un principio como un padecimiento de un solo gen,62 pero es probable que se explique por un grupo heterogéneo de alteraciones genéticas.33 Afecta a uno a dos porciento de la población. En este tipo de hiperlipidemia aumenta la síntesis y secreción de apo B y LMBD. Las LMBD parecen ser normales en su composición, pero las LBD son pequeñas y densas, y tienen una menor relación entre colesterol y apo B. La eficacia de la lipólisis en los individuos afectados es la que determina si la mayor producción de LMBD en el hígado causa concentraciones altas de LMBD, de LBD, o de ambas. Los receptores de LBD parecen ser normales.65 Algunos heterocigotos para la deficiencia de lipasa de lipoproteína satisfacen los criterios para HCF.
Los individuos con HCF suelen tener concentraciones normales de lipoproteínas hasta el inicio de la edad adulta, momento en que pueden presentar uno de tres fenotipos: aumento en la concentración de LMBD, aumento en la concentración de LBD, o elevación de los niveles de ambas. La expresión fenotípica de este padecimiento puede cambiar en un mismo individuo con el paso del tiempo. Por motivos no bien comprendidos, esta enfermedad se asocia con frecuencia con obesidad y diabetes. Los pacientes con HCF no desarrollan xantomas, pero la enfermedad sí se asocia con mayor riesgo de enfermedad coronaria prematura. De hecho, los individuos afectados corresponden aproximadamente al 10 porciento de los hombres menores de 60 años que sobreviven a un infarto del miocardio.
El diagnóstico de HCF se realiza por medio de una historia familiar cuidadosa y examinando el perfil de lipoproteínas en ayuno del paciente y de sus familiares. Debe existir antecedente franco de enfermedad coronaria prematura y alteraciones múltiples de las lipoproteínas, que afectan aproximadamente a la mitad de los parientes en primer grado. En estos pacientes, la concentración de triglicéridos, la de colesterol de LBD, o ambas, estarán por arriba de la percentila 90, y los triglicéridos serán menores a 1,000 mg/dl.
El tratamiento consiste en disminución de peso (cuando es necesaria), control de la diabetes, ejercicio, disminución en la ingesta de ácidos grasos saturados y colesterol, y restricción de alcohol, estrógenos exógenos y otros fármacos que alteren en forma adversa el metabolismo de las LMBD. Si persiste la hiperlipidemia después de seis meses de este tratamiento, se iniciará tratamiento farmacológico. La elección del medicamento depende del fenotipo hiperlipoproteinémico que tenga el paciente.
HIPERCOLESTEROLEMIA FAMILIAR
Etiología y patogenia
La hipercolesterolemia familiar (HF) es un padecimiento autosómico dominante causado por una mutación en el gen que codifica a la proteína receptora de LBD. Brown, Goldstein y colaboradores definieron este padecimiento4 y describieron cinco clases de alelos mutantes que causan deficiencia funcional o absoluta del receptor de LBD.66
Los homocigotos con HF tienen dos alelos mutantes en el locus del receptor para LBD, por lo que tienen una incapacidad total o casi total para eliminar los remanentes de LMBD y las LBD de la circulación por medio de los receptores de LBD. Los individuos heterocigotos tienen un alelo normal y, por lo tanto, la mitad de la actividad normal en los receptores. En vista de que los remanentes de LMBD son depurados del plasma por el receptor de LBD, la deficiencia de estos receptores aumenta la conversión de LDI a LBD, que se acumulan en el plasma por la deficiencia de receptores. A mayor reducción en la actividad de los receptores, mayor elevación de las LBD por mayor producción (conversión a partir de LDI) y menor depuración. Las concentraciones elevadas de LBD provocan la captación (no mediada por receptores) de las mismas en el tejido conectivo y en las paredes arteriales, causando xantomas y ateroesclerosis.
Manifestaciones clínicas
HF homocigota El estado homocigoto ocurre en alrededor de uno en un millón de personas, y existe mayor incidencia de consanguinidad en los padres de los individuos homocigotos. Estos pacientes tienen hipercolesterolemia importante al nacimiento, desarrollan enfermedad coronaria durante las primeras tres décadas de la vida y se han notificado infartos al miocardio hasta alos 18 meses de edad. Los individuos afectados suelen no sobrevivir después de los 30 años de edad. Durante los primeros años de vida desarrollan xantomas cutáneos amarillo-naranja que son característicos de los homocigotos y que se localizan con frecuencia entre los dedos. También presentan xantomas tendinosos, típicamente en el tendón de Aquiles y en los tendones extensores de las manos, y un arco corneal. Pueden tener episodios de poliartritis y tenosinovitis. En ocasiones existen xantomas en las válvulas cardiacas, que causan estenosis aórtica y, con menos frecuencia, insuficiencia o estenosis mitral. También puede desarrollarse estenosis aórtica supravalvular por afección ateromatosa grave de la raíz aórtica. La concentración total de colesterol en los pacientes homocigotos varía entre 600 a 1,200 mg/dl. La concentración de colesterol de LBD es alrededor de seis veces mayor que en individuos normales, la concentración de triglicéridos es normal o sólo ligeramente alta y el nivel de colesterol de LAD suele estar disminuido. Es posible hacer diagnóstico prenatal a partir del líquido amniótico.
HF heterocigota La forma heterocigota de esta enfermedad tiene una prevalencia de uno en 500 personas, lo que la convierte en el padecimiento genético más frecuente. La hipercolesterolemia puede detectarse desde el nacimiento en la sangre del cordón umbilical. En los individuos afectados se desarrolla enfermedad coronaria prematura, manifestándose los síntomas (en los varones) alrededor de la tercera o cuarta década de la vida. Al llegar a los 60 años, por lo menos el 50 porciento de los hombres han tenido un infarto al miocardio; los síntomas en las mujeres suelen presentarse 10 años más tarde. Comienzan a aparecer xantomas tendinosos alrededor de los 20 años de edad, y éstos existen hasta en el 70 porciento de los heterocigotos mayores de 30 años. También el xantelasma (xantomas cutáneos de los párpados) y el arco corneal son comunes después de esa edad. Los xantomas tendinosos son un signo muy específico de HF, pero el xantelasma y el arco corneal pueden existir en individuos con colesterol normal. Las concentraciones de colesterol total varían entre 300 y 500 mg/dl. Los triglicéridos están aumentados en el 10 porciento de los pacientes heterocigotos, y la concentración de colesterol de LAD es menor de lo normal. Alrededor del cinco porciento de los varones que sobreviven a un infarto del miocardio son heterocigotos para la HF.
Diagnóstico
El diagnóstico de HF homocigota suele hacerse por clínica ante la presencia de xantomas cutáneos, hipercolesterolemia grave (concentración de colesterol total > 650 mg/dl) y triglicéridos normales. La función del receptor de LBD puede medirse en laboratorios especiales usando cultivos de fibroblastos o linfocitos circulantes. Debe sospecharse HF heterocigota cuando se detecta hipercolesterolemia grave por aumento en las LBD. Si existen xantomas tendinosos, esto confirma el diagnóstico, si no, deberán investigarse causas de hipercolesterolemia secundaria, aunque no se excluye el diagnóstico de hipercolesterolemia familiar. Una historia clínica cuidadosa puede revelar el antecedente de enfermedad coronaria prematura e hipercolesterolemia, que afecta a alrededor de la mitad de los familiares en primer grado. La existencia de hipercolesterolemia y xantomas tendinosos en un padre o un hijo es virtualmente diagnóstica, lo mismo que la hipercolesterolemia en un niño de la familia. Es muy importante realizar estudios de escrutinio a los familiares en primer grado para poder proporcionarles el tratamiento y orientación adecuados.
Tratamiento
HF homocigota El enfoque terapéutico del paciente homocigoto depende de la presencia o ausencia de actividad del receptor fijador de LBD. Si el paciente no tiene actividad, la administración de un tratamiento que estimule la producción de receptores de LDL no tendrá utilidad (v.gr., tratamiento dietético, resinas fijadoras de ácidos biliares, estatinas y cirugía de derivación ileal). Sin embargo, se ha reportado una reducción de la LBD de 20 porciento con el tratamiento con atorvastatina en pacientes con receptores negativos. Si el paciente tiene alguna función de los receptores, es posible disminuir en forma significativa las LBD combinando resinas fijadoras de ácidos biliares y una estatina. El ácido nicotínico, un fármaco que disminuye la LBD al reducir la síntesis de su precursor, la LMBD, puede ser eficaz para disminuir el colesterol de LBD en individuos homocigotos, independientemente del tipo de defecto de receptor que tengan.
En los homocigotos, la nutrición parenteral total ha demostrado disminuir las concentraciones de LBD,67 lo mismo que la cirugía de derivación portocava. Históricamente, el método terapéutico más empleado en estos pacientes ha sido la eliminación de las LBD por plasmaféresis. Una nueva modificación para este tratamiento es la aféresis, en ella se extrae el plasma del paciente, se elimina en forma selectiva el colesterol de LBD (pero no el colesterol de LAD), y se reinfunde el plasma al enfermo. El método más dramático de tratamiento consiste en el trasplante hepático, que proporciona al paciente receptores hepáticos de LBD y disminuye el nivel de LAD en forma inmediata hasta concentraciones cercanas a lo normal.68 El tratamiento genético directo para administrar al hígado genes normales de receptor de LDL también es un tratamiento potencial que puede ser atractivo para el paciente con HF homocigota.
HF heterocigota El objetivo del tratamiento consiste en disminuir la concentración de colesterol a menos de 130 mg/dl, o menos si el paciente tiene ya enfermedad coronaria. Es posible estimular el gen normal que poseen los pacientes homocigotos, por lo que las resinas que fijan ácidos biliares y las estatinas son eficaces. También es útil el ácido nicotínico. Con frecuencia se requiere tratamiento combinado con dos fármacos, e incluso pueden ser necesarios tres. Aunque la dieta por sí sola no es suficiente para los heterocigotos con HF, el reducir la ingesta de ácidos grasos saturados y colesterol disminuye la concentración de LBD y reduce la cantidad de fármacos requeridos. Estudios realizados en animales han demostrado que las dietas ricas en grasa suprimen la actividad del receptor de LBD.4 Otra forma de estimular la síntesis de receptores de LBD es evitando que las sales biliares regresen al hígado por medio de una cirugía de derivación ileal, pero este tratamiento es un poco obsoleto con la disponibilidad de medicamentos reductores de la LBD. Se ha demostrado que los xantomas tendinosos desaparecen cuando la concentración de LBD se mantiene en un rango adecuado. La reducción agresiva del colesterol de la LBD en hombres y mujeres que tienen hipercolesterolemia familiar heterocigota puede inducir regresión de la ateroesclerosis coronaria.69
DEFECTO FAMILIAR EN LA APOLIPOPROTEINA B-100
La apo B-100 defectuosa es otra causa genética de elevación en la concentración de LBD. No se conoce la prevalencia de esta enfermedad, pero se calcula que es de uno en 500 a 700 personas. La estructura y función de los receptores de LBD es normal en estos pacientes, y se produce y secreta una apo B-100 de longitud normal, pero que se fija en forma defectuosa a los receptores, por lo que se acumula LBD en el plasma. Los individuos afectados tienen un cuadro clínico que puede ser indistinguible del de la hipercolesterolemia familiar heterocigota: tienen hipercolesterolemia grave, pueden formar xantomas tendinosos, y desarrollan ateroesclerosis prematura. Al parecer, el tratamiento con estatinas disminuye la concentración de colesterol de LBD en los enfermos con este padecimiento.70 Se requieren pruebas especializadas, disponibles solo en laboratorios de investigación, para identificar a los individuos afectados.
HIPOBETALIPOPROTEINEMIA Y ABETALIPOPROTEINEMIA FAMILIARES
La hipobetalipoproteinemia familiar puede ser causada por diferentes mutaciones del gen de la apo B, que interfieren con la traslocación de la molécula completa y funcional de apo B-100. Se requiere que la molécula esté completa para la síntesis y secreción de LMBD en el hígado, por lo que a diferencia de los pacientes con apo B-100 defectuosa, los heterocigotos suelen estar asintomáticos, tienen concentraciones de colesterol de LBD de un cuarto a un tercio de lo normal y su riesgo de enfermedad coronaria es menor.3 Los homocigotos tienen diversos síntomas, y los más afectados son difíciles de distinguir de los enfermos con abetalipoproteinemia.
La abetalipoproteinemia es un síndrome autosómico recesivo poco frecuente en el cual la apo B se sintetiza en forma normal, pero en donde la síntesis y secreción de las lipoproteínas que la contienen están alteradas. Los individuos afectados tienen concentraciones muy bajas de colesterol y triglicéridos, esteatorrea, acantosis, retinopatía degenerativa y enfermedad neuromuscular progresiva.
DISBETALIPOPROTEINEMIA
La disbetalipoproteinemia, también llamada hiperlipoproteinemia tipo III y enfermedad de franja beta ancha, se define como la presencia de partículas de LMBD que migran hacia la posición beta en la electroforesis (en condiciones normales, las partículas de LMBD migran hacia la posición pre-beta). Las partículas de LMBD-beta están formadas por quilomicrones y remanentes de LMBD. La disbetalipoproteinemia está causada en parte por una apo E mutante que impide la captación hepática de las lipoproteínas que la contienen y que disminuye la conversión de LMBD a LDI y LBD. Sin la presencia de algún defecto genético, hormonal o ambiental adicional, los remanentes no se acumulan en grado suficiente como para causar hiperlipidemia porque son depurados por receptores hepáticos que también se fijan, aunque con menos avidez, a las apo B-48 y apo B-100. La disbetalipoproteinemia se presenta cuando el defecto de la apo E se combina con algún defecto genético o adquirido que causa sobreproducción de LMBD (como la obesidad o la HCF) o disminución en la actividad de los receptores de LBD, como la HF heterocigota.60
Existen tres alelos comunes para la apo E: E2, E3 y E4. Estos alelos se encuentran en cualquiera de seis posibles combinaciones. La apo E3 y apo E4 se unen con más avidez al receptor de LBD que la apo E2, que es la forma mutante de apo E responsable de este padecimiento. El genotipo E2/E2 se encuentra en el uno porciento de la población caucásica, y virtualmente en todos los individuos con disbetalipoproteinemia. Se desarrolla hiperlipoproteinemia en sólo uno porciento de las personas con genotipo E2/E2. Se han descrito otras mutaciones raras de la apo E, que parecen producir el mismo síndrome que la homocigocidad E2/E2.71
Los individuos con disbetalipoproteinemia tiene aumento tanto en el colesterol como en los triglicéridos, y tienen propensión para desarrollar enfermedad coronaria prematura. Por motivos no conocidos, estas personas tienen también mayor riesgo de enfermedad vascular periférica. La hiperlipidemia no se presenta antes de la edad adulta. Los xantomas palmares (xantoma astriata palmaris), coloración amarillo-naranja de los surcos de la palma, son patognomónicos de la disbetalipoproteinemia, pero no siempre están presentes. Con frecuencia se observan xantomas tuboeruptivos en sitios de presión, sobre todo en codos, glúteos y rodillas. Pueden existir xantomas tendinosos, el xantelasma es raro. Suele asociarse obesidad, y la diabetes o el hipotiroidismo pueden enmascarar la enfermedad.
Debe sospecharse diagnóstico de disbetalipoproteinemia en cualquier individuo con aumento del colesterol total y de los triglicéridos, aumento en la concentración de colesterol de LMBD, y disminución en la concentración de colesterol de LBD y LAD. Los niveles de colesterol y triglicéridos varían de 300 a 500 mg/dl y son más o menos equivalentes, excepto durante las exacerbaciones agudas, cuando predomina la hipertrigliceridemia. En la electroforesis se observa migración beta de las LMBD, y la ultracentrifugación demuestra que la relación entre colesterol de las LMBD y triglicéridos de las mismas es mayor de 0.3. El diagnóstico definitivo se realiza al detectar el genotipo E2/E2 por enfoque isoeléctrico. Deberá excluirse la presencia de diabetes e hipotiroidismo.
El tratamiento de la disbetalipoproteinemia es básicamente el mismo de las otras hipertrigliceridemias: reducir el peso hasta el ideal, optimizar el control de la glucosa si coexiste diabetes y evitar el alcohol, estrógenos y otros fármacos que aumenten los triglicéridos. La dieta debe ser baja en grasas totales, grasas saturadas, colesterol y carbohidratos simples. Los pacientes deben realizar ejercicio en forma regular. Si la hiperlipidemia no mejora con estas medidas, el gemfibrozil, el ácido nicotínico (niacina) y las estatinas han demostrado ser eficaces. Por lo general, la disbetalipoproteinemia responde con facilidad al tratamiento, y se ha observado regresión de los xantomas.
SINDROMES PRIMARIOS DE BAJA CONCENTRACION DE LAD
La concentración baja de LAD puede ser causada por menor síntesis, mayor catabolismo o ambos. Es probable que la menor síntesis facilite la ateroesclerosis, pero el catabolismo rápido no. En muchos de los trastornos primarios de baja concentración de LAD no se ha definido bien el metabolismo de la LAD. La deficiencia de aciltransferasa de lectina-colesterol es una enfermedad autosómico recesiva que es consecuencia de la ausencia de aciltransferasa de lecitina-colesterol o de la presencia de una forma inactiva de esta enzima en el plasma de los homocigotos afectados. Los heterocigotos tienen disminuciones leves de las LAD. En la forma homocigota no existe LAD2 madura porque el colesterol libre no puede ser esterificado ni transportado hacia el núcleo de la LAD3. Sólo existen formas tempranas de LAD, y su concentración suele ser de alrededor de 5 mg/dl. Los niveles de LMBD y LBD son normales. Los homocigotos sufren anemia, opacificación corneal e insuficiencia renal. Se ha reportado enfermedad coronaria prematura.
La enfermedad de ojo de pescado es un padecimiento poco frecuente causado por deficiencia parcial de la aciltransferasa de lecitina-colesterol. Los pacientes tienen LAD bajas, elevaciones moderadas de los triglicéridos (250 a 350 mg/dl), LMBD y LBD ricas en triglicéridos y opacificación corneal severa.72
La enfermedad de Tangier es un padecimiento autosómico recesivo causado por un defecto molecular desconocido que provoca deficiencia severa o ausencia de LAD. Las concentraciones de triglicéridos son normales o elevadas, y se acumula éster colesteril en los tejidos. Entre las manifestaciones clínicas destacan hipertrofia amigdalina de color naranja, esplenomegalia y neuropatía periférica. A pesar de que los niveles de LAD son muy bajos, la frecuencia de enfermedad coronaria no está aumentada en estos pacientes.
Los defectos moleculares de la apo A-I ocasionan un nivel bajo de LAD. Cuando estos defectos son aislados no siempre se asocian con un mayor riesgo de ateroesclerosis. La apo A-IMilano es una forma mutante de apo A-I que se asocia con concentraciones muy bajas de LAD (7 a 14 mg/dl) y niveles altos de triglicéridos, pero no con mayor riesgo de enfermedad coronaria.73 La mutación causa mayor afinidad de la apo A-I por el colesterol, causando catabolismo acelerado y mayor captación del mismo en los tejidos.74 Una enfermedad genética diferente con deficiencia severa de apo A-I y ausencia de apo C-III causa xantomas tendinosos, opacificación corneal y enfermedad coronaria prematura.75
Es frecuente que las causas primarias de hipertrigliceridemia causen disminución en la concentración de LAD. Como se analizó antes, se requiere de la lipólisis de las lipoproteínas ricas en triglicéridos para la producción de partículas maduras de LAD. La existencia de un padecimiento específico y monogénico de hipoalfalipoproteinemia familiar es motivo de controversia. Este término ha sido usado para describir a miembros de familias con concentraciones de LAD que se encuentran en la percentila 10, con o sin hipertrigliceridemia asociada.
HIPERCOLESTEROLEMIA POLIGENICA
La mayoría de los individuos con concentración de colesterol por arriba de la percentila 95 no tienen una causa monogénica o secundaria identificable, y caen dentro de la categoría mal definida de hipercolesterolemia poligénica.62 Estos pacientes se distinguen de los heterocigotos con HF o HCF porque no tienen antecedentes familiares de enfermedad cardiovascular prematura. Cuando se conozca más en relación con la biología molecular de la hipercolesterolemia, podrá encontrarse que estos individuos tienen una o más alteraciones específicas, como aumento en la absorción de colesterol, defectos leves en la función de los receptores de LBD o en la estructura de la apo B, o reducción del número de receptores de LBD por la interacción de múltiples causas genéticas o ambientales. Después de descartar causas secundarias, estos pacientes deben recibir el tratamiento estándar para el aumento en el colesterol de LBD .
Alteraciones secundarias del metabolismo de los lípidos
Las disbetalipoproteinemias secundarias son causadas por defectos adquiridos del metabolismo de los lípidos que causan hipercolesterolemia, hipertrigliceridemia, o hiperlipidemia combinada, con o sin disminución del nivel de LAD [ver tabla 4]. La hipertrigliceridemia secundaria puede ser lo suficientemente grave como para causar quilomicronemia. Cuando la dislipoproteinemia es originada por un medicamento, éste deberá suspenderse siempre que sea posible. Las dislipoproteinemias secundarias causan las mismas alteraciones que las dislipoproteinemias primarias, esto es, ateroesclerosis y pancreatitis.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Puede asociarse con quilomicronemia si existe algún padecimiento primario concomitante que cause hipertrigliceridemia. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
La diabetes mellitus causa aumento en la síntesis y reducción en el catabolismo (debido a disminución en la actividad de la lipasa de lipoproteína) de las LMBD, lo que produce hipertrigliceridemia y disminución en la concentración de LAD. Las LBD están normales o aumentadas. Se produce quilomicronemia en ayuno cuando existe alguna forma primaria o secundaria de hipertrigliceridemia. Las LMBD y los quilomicrones compiten en su unión a la lipasa de lipoproteína, y pueden acumularse ambas lipoproteínas. La disminución en las LAD es consecuencia de la menor lipólisis de las lipoproteínas ricas en triglicéridos, que proporcionan componentes para la síntesis de LAD. La dislipoproteinemia se presenta tanto en la diabetes insulinodependiente como en la no insulinodependiente. Las concentraciones de lípidos deben normalizarse al tratar la diabetes, si esto no sucede deberán buscarse causas adicionales. En los diabéticos con hipertrigliceridemia persistente, el gemfibrozil es adecuado porque aumenta la actividad de la lipasa de lipoproteína y quizá reduce la secreción de LMBD en el hígado. Por lo general debe evitarse el ácido nicotínico, en especial en diabéticos no insulinodependientes, porque aumenta la hiperglucemia.76 Las estatinas también son eficaces para la dislipemia de la diabetes.76
El hipotiroidismo causa un aumento severo de las LBD por disminuir la actividad del receptor, y con frecuencia provoca hipertrigliceridemia con reducción asociada de las LAD secundaria a una menor actividad de la lipasa de lipoproteínas.77 También pueden acumularse remanentes de quilomicrones y LMBD, causando una disbetalipoproteinemia de tipo III. La dislipoproteinemia del hipotiroidismo se corrige con el tratamiento sustitutivo de hormonas tiroideas.
El síndrome nefrótico causa aumento en la secreción hepática de las liproteínas que contienen apo B-100 (i.e., LMBD) en respuesta a la pérdida de albúmina y otras proteínas en la orina. La síntesis hepática de colesterol también esta aumentada, y esto puede disminuir el número de receptores hepáticos de LBD y la depuración de las LBD circulantes. Existe un aumento severo de la concentración de LBD y, en las etapas tardías del padecimiento pueden aumentar también las LMBD, lo que se asocia a un nivel bajo de LAD por la alteración de la lipólisis.78 Los pacientes con síndrome nefrótico tienen mayor riesgo de enfermedad coronaria debido a la hipercolesterolemia, y la alteración de los lípidos debe tratarse en forma agresiva cuando el pronóstico general del paciente es bueno. La dieta, la pérdida de peso y el ejercicio pueden mejorar las concentraciones de lipoproteínas, pero casi siempre se requieren medicamentos para alcanzar los niveles ideales. Las resinas fijadoras de ácidos biliares son eficaces para disminuir la concentración de LBD en los enfermos nefróticos, pero si se utilizan como agentes únicos, no disminuyen las LBD lo suficiente y pueden aumentar la hipertrigliceridemia. El ácido nicotínico debe ser útil porque inhibe la secreción hepática de las lipoproteínas que contienen apo B-100; sin embargo, su uso no ha sido estudiado en forma suficiente. Las estatinas pueden ser eficaces en los pacientes con síndrome nefrótico, aunque tampoco existe mucha experiencia respecto a su uso. Suele requerirse un tratamiento combinado para reducir la concentración de LBD, y la más eficaz es la de una resina más una estatina.
La insuficiencia renal crónica produce hipertrigliceridemia como resultado de la deficiencia de lipasa de lipoproteínas (LLP) y de la lipasa hepática de triglicéridos.78 Las concentraciones de triglicéridos suelen ser de 200 a 750 mg/dl, y las de LAD son bajas. El riesgo de enfermedad coronaria es mayor que el de la población general. Debe probarse el tratamiento no farmacológico antes de considerar administrar medicamento, a menos de que la concentración de triglicéridos sea mayor a 1,000 mg/dl. El tratamiento farmacológico de la hipertrigliceridemia en la insuficiencia renal crónica es motivo de controversia por la diferencia de opiniones en relación con el papel de la hipertrigliceridemia en la aterogénesis. Se ha demostrado que el gemfibrozil, un medicamento que aumenta la actividad de la lipasa de lipoproteína, es útil en estos casos. En vista de que se excreta por vía renal, la dosis debe reducirse para evitar aumentar el riesgo de miopatía. El uso de ácido nicotínico y estatinas en casos de insuficiencia renal crónica no ha sido aún bien estudiado.
En sus etapas tempranas la cirrosis biliar primaria causa elevación leve de las LMBD y LBD, y aumento importante de las LAD. Al empeorar la enfermedad existe aumento severo de colesterol por la regurgitación de bilis hacia el plasma, lo que crea una partícula anormal de lipoproteína que es rica en colesterol libre y fosfolípidos, llamada lipoproteína X. La alteración en la actividad hepática de la lipasa de triglicéridos y de la esterificación del colesterol contribuye a la dislipoproteinemia. La lipoproteína X puede ser causada también por otras formas de ictericia obstructiva.
El consumo de alcohol aumenta la producción hepática de triglicéridos al aumentar la disponibilidad de ácidos grasos libres por varios mecanismos.79 Los ácidos grasos en exceso son incorporados a triglicéridos, que a su vez se agregan a las partículas de LMBD, que son secretadas en mayor cantidad. Si la actividad de la LLP se altera por algún padecimiento coexistente, las LMBD se acumulan y compiten con los quilomicrones por los sitios de la unión de la lipasa de lipoproteína, provocando quilomicronemia.77 El alcohol también aumenta la concentración de LAD, y este efecto puede explicar la asociación entre el uso moderado y regular del alcohol con un menor riesgo de enfermedad coronaria. Existen estudios que han demostrado que la LAD2, la LAD3, o ambas subfracciones, aumentan con el consumo de alcohol,77 lo mismo que la concentración de apo A-1. El alcohol no tiene efectos significativos sobre las concentraciones de LBD.
Los anticoncepticos orales que contienen combinaciones de estrógenos y progesterona tienen efectos variables sobre las lipoproteínas, dependiendo de la combinación específica. Los estrógenos tienden a aumentar las LMBD y LAD, y a disminuir las LBD. Los progestágenos tienden a disminuir las LAD y aumentar las LBD, pero su efecto varía en forma muy importante. La sustitución posmenopáusica de estrógenos disminuye la LBD y aumenta la LAD, la adición de progesterona para proteger el útero disminuye estos efectos, pero no los elimina.80 Los estrógenos también aumentan los niveles de triglicéridos, en especial en mujeres con sobrepeso, lo que obliga a vigilarlos.
Enfoque práctico para los trastornos comunes
La mayoría de los pacientes con un trastorno de los lípidos tienen uno de tres patrones generales: predominio de aumento en el colesterol causado por elevación en la concentración de LBD, una hiperlipidemia combinada con aumento de triglicéridos y colesterol, o elevación predominante de los triglicéridos con nivel normal o bajo de LBD. En esta sección se analizará también un trastorno menos frecuente, la reducción aislada de colesterol de LAD.
PACIENTES CON AUMENTO DEL COLESTEROL
Los pacientes con concentración elevada de colesterol tienen una concentración de colesterol de LBD mayor de 130 mg/dl, pero nivel de triglicéridos normales (< 200 mg/dl). La concentración de colesterol de LAD es variable, pero puede ser normal. Los trastornos de los lípidos en estos pacientes suelen descubrirse por escrutinio de rutina. Aunque algunos observadores han puesto en duda la relación costo-eficacia de realizar escrutinio a varones y mujeres jóvenes en busca de hipercolesterolemia,81 la gran prevalencia del aumento en el colesterol asociado a LBD y total en plasma en los Estados Unidos justifica realizar estudios de escrutinio en la población, según lo recomiendan el NCEP y otras autoridades. Este tipo de escrutinio también es adecuado si el paciente tienen alguna preocupación especial sobre su estado de riesgo o si existen antecedentes familiares de enfermedad cardiovascular a edades tempranas o alteraciones de lípidos. El tratamiento de los pacientes con concentraciones de colesterol total por debajo de 240 mg/dl y que no tienen otros factores de riesgo de enfermedad cardiovascular deben consistir en modificar la dieta, el peso corporal y realizar ejercicio.
Intervención dietética
Con frecuencia los médicos se sienten frustrados cuando aconsejan a sus pacientes cambiar su dieta, porque esta medida produce cambios muy pequeños en la concentración de colesterol en muchos pacientes. En la experiencia de los autores, el colesterol de LBD sí puede mejorar mucho con la dieta y el cambio en el estilo de vida, dependiendo en parte de la dieta basal y el grado de cambio. A pesar de ello, muchos pacientes tienen dificultades para controlar su peso y mantener una dieta baja en grasas y un programa regular de ejercicio. Antes de optar por el tratamiento farmacológico, el médico debe considerar varios factores, sobre todo el riesgo cardiovascular global del individuo y los riesgos conocidos y desconocidos del tratamiento farmacológico. El riesgo puede cuantificarse usando los niveles de los factores de riesgo a partir de los resultados de estudios epidemiológicos.82 También debe considerarse que las variaciones de laboratorio pueden ocultar las reducciones pequeñas en la concentración de colesterol, que una disminución del cinco porciento en el nivel de LBD disminuye el riesgo de enfermedad coronaria en un 10 porciento (un efecto significativo), y que el seguimiento regular puede ayudar a maximizar los cambios a largo plazo en el estilo de vida. Con frecuencia es adecuado conformarse con un nivel más alto de colesterol que el ideal si los riesgos del siguiente nivel de tratamiento sobrepasan los beneficios probables. Los médicos deben insistir en la dieta y el ejercicio, y pueden ayudar a disminuir el riesgo de enfermedad cardiovascular en la población sin prescribir medicamentos en forma excesiva.
Las normas del NCEP indican modificar la dieta si el nivel de colesterol de las LBD es mayor de 160 mg/dl (130 mg/dl si se asocian otros dos factores de riesgo) y administrar tratamiento farmacológico si la concentración es mayor de 190 mg/dl después de la modificación en la dieta (160 mg/dl en presencia de otros dos factores de riesgo). En presencia de enfermedad vascular conocida, el objetivo debe ser lograr una concentración de colesterol de LBD de 100 mg/dl. Estas cifras son razonables, pero los autores recomiendan individualizar las cifras límite en cada paciente.82 Por ejemplo, la concentración de colesterol de LBD tiende a aumentar con la edad, por lo que los límites deben ser menores en los pacientes jóvenes.83 Sin embargo, el tratamiento farmacológico es inapropiado para la mayoría de los varones jóvenes y mujeres premenopáusicas que tienen concentraciones de colesterol de LBD por debajo de 220 mg/dl y no tienen otros factores de riesgo. La presencia de comorbilidad importante (v.gr., enfermedad pulmonar obstructiva crónica grave o insuficiencia cardiaca) debe moderar la agresividad del médico, lo mismo que un aumento marcado en la concentración de colesterol de LAD (v.gr., > 70 mg/dl).84 Una concentración de colesterol de LAD por debajo de 35 mg/dl justifica esfuerzos más intensos para disminuir la concentración de LBD, así como el aumento de la Lp(a).
La mayoría de los pacientes con aumento en la concentración de colesterol de LBD no tienen un padecimiento primario de los lípidos, la elevación refleja sólo la susceptibilidad del metabolismo de lípidos en el humano a la ingesta de grasas saturadas y colesterol, a la obesidad y a la falta de ejercicio. Puede sospecharse un padecimiento primario si existe una clara historia familiar de enfermedad ateroesclerótica o alteraciones de lípidos, o si se encuentran xantomas tendinosos, pero esta diferencia tiene poca utilidad terapéutica, por lo menos al principio. También hay que tener en cuenta las causas de hipercolesterolemia secundaria [ver tablas 4 y 5], aunque la mayoría son evidentes, y se aconseja realizar pruebas de detección de hipotiroidismo.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
* Puede asociarse con quilomicronemia si coexiste alguna alteración primaria o secundaria
|
El tratamiento inicial para el aumento en el colesterol de LBD consiste en disminuir las grasas saturadas y el colesterol de la dieta; con frecuencia es útil la ayuda de un nutriólogo, así como el material escrito de diferentes tipos. Algunos pacientes están muy motivados para modificar su dieta (con objeto de evitar los medicamentos), y una dieta con un contenido total de grasa de menos del 20 porciento de las calorías suele disminuir la concentración de colesterol de LBD en la mayor parte de los casos. Otros pacientes tiene menos interés, pero se recomienda intentar la modificación dietética por lo menos durante seis meses antes de iniciar tratamiento farmacológico, a menos de que el paciente tenga enfermedad coronaria diagnosticada o una concentración de colesterol de LBD que exceda en forma constante los 200 mg/dl. La reducción de grasas en la dieta disminuye tanto el colesterol de LAD como el de LBD,85 pero la reducción en la LAD es causada por aumento en el catabolismo de estas lipoproteínas, lo que puede aumentar el transporte inverso de colesterol.86 No se sabe que la disminución de las LAD por la dieta sea dañina.87 El control de peso y el ejercicio pueden minimizar la reducción o incluso aumentar la concentración de LAD en los pacientes con una dieta baja en grasas.85
Tratamiento farmacológico
Las resinas fiadoras de ácidos biliares y las estatinas son los medicamentos que se recomiendan de inicio para disminuir el colesterol de LBD. Los autores prefieren las resinas en pacientes jóvenes y en quienes tienen indicaciones limítrofes para usar medicamentos por su poca toxicidad potencial. Sin embargo, la constipación y las molestias consecuentes hacen que estos fármacos sean poco tolerados, por lo que el médico deberá animar al paciente para que los use. La constipación puede mejorar si se agrega fibra a la dieta (en especial cereal de avena88) o una preparación de psyllium,89 además de que ambos contribuyen a disminuir la LBD. Con frecuencia las estatinas tienen mejor costo-beneficio y se asocian con mayor cumplimiento del paciente que las resinas, además de que se ha demostrado que disminuyen la mortalidad total.19 Por lo tanto, está justificado emplearlas en pacientes de mayor riesgo (v.gr., en prevención secundaria) y en enfermos con historia de constipación u otras contraindicaciones relativas al tratamiento con resinas. Las estatinas también disminuyen las concentraciones de triglicéridos y aumentan en forma modesta los niveles de LAD. La niacina también es muy eficaz para disminuir el colesterol de LBD, pero por sus muchos efectos adversos suele reservarse para pacientes con hiperlipidemia combinada, nivel bajo de LAD o ambos, o para pacientes que no toleran los otros dos tipos de fármacos. Muchos pacientes requieren de tratamiento combinado para lograr concentraciones de colesterol de LBD menores de 130 mg/dl (100 mg/dl para los pacientes con enfermedad vascular conocida), las resinas y las estatinas son sinérgicas en su efecto sobre el colestrol de LBD, y la niacina puede combinare con resinas, estatinas o ambas (con cierta precaución por la asociación con miositis al usar estatinas).
PACIENTES CON HIPERLIPIDEMIA COMBINADA
En la hiperlipidemia combinada, tanto la concentración de colesterol como la de triglicéridos está elevada. Típicamente la concentración de triglicéridos varía entre 200 y 800 mg/dl, la de LBD es mayor de 100 mg/dl y la de LAD menor de 40 mg/dl. La hiperlipidemia combinada parece incluir varios trastornos genéticos que están mal caracterizados. Algunos pacientes simplemente muestran dos trastornos frecuentes simultáneos, la elevación poligénica de colesterol de LBD y la hipertrigliceridemia inducida por sobrepeso. Otros enfermos pueden tener hiperlipidemia familiar combinada, que se diagnostica solo por medio de estudios familiares.33 Otro subgrupo de pacientes muestra varios factores de riesgo cardiovasculares que incluyen obesidad central, hiperinsulinemia, hipertensión y las alteraciones de lípidos consistentes en elevación de triglicéridos y reducción del colesterol de LAD.90 Los pacientes afectados parecen tener niveles elevados de partículas de LBD pequeñas y densas,32 que son susceptibles a la oxidación y pueden ser muy aterogénicas. Los pacientes con hiperlipidemia combinada parecen tener mayor riesgo de enfermedad cardiovascular, pero existen pocos estudios intervencionistas dirigidos a este subgrupo de pacientes.
La evaluación inicial incluye la medición de otros factores de riesgo cardiovascular y una historia familiar detallada. La presencia de enfermedad cardiovascular prematura en la historia familiar es un marcador importante de mayor riesgo. El escrutinio de estos pacientes en busca de causas secundarias de trastornos de lipoproteínas [ver tablas 4 y 5] es importante antes de iniciar el tratamiento.
Cambios de comportamiento
El ejercicio y la pérdida de peso son las intervenciones de comportamiento más importantes porque la mayoría de los pacientes con hiperlipidemia combinada tienen exceso de peso y porque inclusive las pérdidas pequeñas de grasa corporal (3 a 6 kg) disminuyen en forma importante el nivel de triglicéridos, y con frecuencia producen un incremento pequeño en la concentración de LAD.91 Por supuesto, los pacientes con factores de riesgo como hiperinsulinemia o hiperglucemia e hipertensión se beneficiarán aún más en todos los factores de riesgo por la pérdida de peso inducida por el ejercicio. Por lo tanto, el médico debe hacer su mayor esfuerzo para convencer al paciente de aumentar el nivel de ejercicio e intentar disminuir de peso. Las visitas frecuentes y el seguimiento telefónico pueden ser útiles para el mayor cumplimiento por parte del paciente.
Debe restringirse la grasa saturada en la dieta en los pacientes con hiperlipidemia familiar, y esta restricción puede ayudar a disminuir la concentración de colesterol de LBD. Sin embargo, algunos pacientes presentan exacerbación de su hipertrigliceridemia si reducen su ingesta total de grasa en la dieta a menos de 25 a 30 porciento de calorías. Estos pacientes pueden beneficiarse por aumentar la ingesta de grasas no saturaras (i.e., grasas vegetales) en un grado moderado, aunque cuando la dieta baja en grasa se asocia con pérdida de peso, es raro que aumente la concentración de triglicéridos.
Tratamiento farmacológico
Existen pocos estudios clínicos para guiar el tratamiento farmacológico de la hiperlipidemia combinada, y la mayoría de éstos se enfocan a la elevación en el colesterol de LBD. El Helsinky Heart Study incluyó hombres con concentraciones de colesterol no de LAD mayor de 200 mg/dl y mostró beneficio del gemfibrozil sobre las tasas de enfermedad cardiovascular, pero no sobre la mortalidad total.17 En este estudio, los pacientes con hiperlipidemia combinada parecieron beneficiarse del tratamiento en forma semejante a otros pacientes,47 pero el número de enfermos con hiperlipidemia combinada fue relativamente pequeño y estos análisis a posteriori pueden ser equívocos. Los autores consideran adecuado emplear medicamentos en los pacientes que son candidatos al mismo por su concentración de colesterol de LBD y en los que tienen enfermedad cardiovascular conocida, varios otros factores de riesgo cardiovasculares o una historia familiar clara de enfermedad cardiovascular prematura. El tratamiento de elección para la hiperlipidemia combinada es la niacina, que produce cambios benéficos en todas las fracciones de lípidos, en especial al disminuir el colesterol de LBD y quizá disminuyendo en forma selectiva el colesterol de LBD de partículas densas y pequeñas (una excepción importante es que la niacina puede empeorar el metabolismo de la glucosa en pacientes diabéticos).92 Las estatinas también pueden ser útiles en este trastorno si la concentración de triglicéridos es menor de 200 mg/dl, ya que causan reducción modesta en la concentración de triglicéridos, pequeño incremento en el nivel de LAD y reducción en el nivel de LBD (la atorvastatina es la estatina más potente para disminuir el nivel de triglicéridos). Las resinas pueden ser útiles en la hiperlipidemia combinada como tratamiento adyuvante para las concentraciones elevadas de colesterol de LBD después de que se ha controlado el nivel de triglicéridos. Por último, los fibratos reducen el nivel de triglicéridos y aumentan el de LAD, pero su efecto sobre el colesterol de LBD es variable.
PACIENTES CON AUMENTO EN LOS TRIGLICERIDOS
Los pacientes con triglicéridos elevados se distinguen de las personas con hiperlipidemia combinada por sus niveles de triglicéridos más altos (con frecuencia > 1,500 mg/dl) y colesterol de LBD normal (< 100 mg/dl y con frecuencia < 80 mg/dl). Los niveles de colesterol de LAD casi siempre son bajos. Este patrón de alteración de lípidos incluye a la hipertrigliceridemia familiar y a otros padecimientos poco frecuentes. Los enfermos pueden tener mayor riesgo de pancreatitis, aunque según los autores muchos pacientes presentan una historia larga de elevación de triglicéridos y no pancreatitis. Algunos pacientes con hipertrigliceridemia tienen diabetes, y es adecuado investigar la glucemia, aunque la diabetes no causa elevaciones importantes en los triglicéridos a menos que exista cetoacidosis franca (i.e., en la diabetes mellitus dependiente de insulina). Deben considerarse otras causas secundarias de triglicéridos elevados [ver tablas 4 y 5]. La historia familiar puede ser útil para evaluar el riesgo individual de enfermedad cardiovascular.
Muchos pacientes con hipertrigliceridemia son obesos y sedentarios, y se benefician del ejercicio y la pérdida de peso. Como en el caso de la hiperlipidemia mixta, los beneficios de la pérdida de peso son tan importantes que el médico debe ser insistente para lograr que el paciente acepte y cumpla los cambios dietéticos y el programa de ejercicio. La ingesta de alcohol debe reducirse o eliminarse.
Los pacientes que son incapaces de perder suficiente peso como para mantener sus concentraciones de triglicéridos por debajo de 500 a 750 mg/dl tienen riesgo de hipertrigliceridemia que, en concentraciones por arriba de 3,000 mg/dl puede producir pancreatitis e incluso hiperviscosidad. Por lo tanto, la mayoría de los especialistas recomiendan el tratamiento farmacológico para esos pacientes. Además, los autores dan tratamiento a los pacientes que no pierden peso suficiente y tienen historia familiar de enfermedad cardiovascular o una concentración de colesterol de LAD menor de 35 mg/dl. Tanto la niacina como el gemfibrozil suelen controlar el nivel de triglicéridos y elevar el de LAD. El gemfibrozil puede aumentar la concentración de LBD, y la niacina debe evitarse en los pacientes diabéticos. Si el gemfibrozil eleva la concentración de LBD por arriba de 130 mg/dl, la niacina puede sustituirse por una resina o puede agregarse una estatina. Existe preocupación sobre la seguridad a largo plazo de los fibratos.93 Una tercera opción farmacológica para la hipertrigliceridemia es el suplemento con ácidos grasos omega-3, pero no se conocen del todo la seguridad a largo plazo y el impacto sobre los puntos clínicos finales.
PACIENTES CON BAJA CONCENTRACION DE COLESTEROL DE LAD
No es claro cual debe ser el tratamiento adecuado de los individuos asintomáticos que tienen niveles bajos de LAD acompañados de concentraciones de LBD y colesterol total normales.94 Hasta que se notifiquen los resultados de los estudios clínicos de prevención que se realizan en la actualidad, el valor del tratamiento farmacológico para elevar los niveles de LAD en este grupo de pacientes es incierto.
En los pacientes con enfermedad coronaria la meta debe ser disminuir la concentración de LBD a menos de 100 mg/dl, o una reducción de más del 30 porciento de la basal, aunque sea menor, independientemente del nivel de LAD. En dos estudios de prevención secundaria, los pacientes con niveles basales bajos de LAD se beneficiaron con el tratamiento con estatinas en la misma forma que los que tenían niveles de LAD más altos.20,44 Las evidencias de los estudios angiográficos sugieren que el aumento en la concentración de LAD puede inhibir la progresión o inducir regresión de la ateroesclerosis coronaria.41,42 En los pacientes con enfermedad coronaria y un nivel bajo de LAD deben intentarse primero medidas no farmacológicas para aumentar esta lipoproteína. Si esto falla deberá considerarse el tratamiento farmacológico, aunque no es claro qué medicamento debe elegirse.95 La colestiramina y el gemfibrozil aumentan la concentración de LAD en plasma al incrementar la síntesis de partículas de LAD (y de apoA-I),96,97 que al parecer facilitan el transporte inverso de colesterol.95 El ácido nicotínico, que ha sido empleado en varios estudios angiográficos, disminuye el catabolismo de las LAD98 y reduce la concentración de LBD en mayor grado que el gemfibrozil. Las estatinas también reducen los niveles de LBD en forma significativa, pero aumentan los de LAD menos que el ácido nicotínico, aunque al igual que el ácido nicotínico, pueden aumentar la síntesis de LAD.99 A menos que la concentración de LBD sea menor de 90 a 100 mg/dl, los autores prefieren el uso de ácido nicotínico en estos pacientes. Cuando los enfermos no lo toleran, usan una estatina, con o sin colestiramina. En los pacientes con niveles de LBD aceptables, el gemfibrozil es una buena opción terapéutica. En todos los casos deben analizarse con el paciente las opciones de tratamiento.
PACIENTES CON ENFERMEDAD CORONARIA Y LIPOPROTEINAS NORMALES
En ocasiones los pacientes tienen enfermedad coronaria pero ninguna alteración de lipoproteínas u otros factores de riesgo. Sin embargo, debe tenerse cuidado antes de concluir que un paciente hospitalizado con enfermedad coronaria tiene una concentración de LBD baja porque ésta disminuye durante un infarto agudo del miocardio y se reduce en forma dramática cuando los pacientes son sometidos a derivación cardiopulmonar. Los autores también han observado disminución importante en la concentración de LBD cuando los pacientes con dolor torácico (en los que se ha descartado un infarto del miocardio), ayunan, se mantienen en posición supina o reciben hidratación y heparina por vía intravenosa. Por lo general es adecuado obtener los valores previos de lipoproteínas cuando el paciente era externo o medir las lipoproteínas en ayuno cuatro a seis semanas después del infarto, en lugar de confiar en los valores durante la hospitalización para el diagnóstico y el tratamento.
Cuando se identifica a un paciente con riesgo aparentemente bajo, debe considerarse investigar concentraciones elevadas de Lp(a) y de homocisteína. Las concentraciones elevadas de Lp(a) pueden aumentar el riesgo de enfermedad coronaria, y esto puede ser totalmente independiente de la concentración de otras proteínas. En pacientes de ascendiencia europea, un nivel mayor de 20 mg/dl se considera alto,57 y si se encuentra, debe considerarse el tratamiento con niacina o, en las mujeres posmenopáusicas sin contraindicaciones, con estrógenos. El valor predictivo de la Lp(a) en otros grupos étnicos requiere de estudio adicional, pero los niveles por arriba de la percentila 80 pueden considerarse elevados.57 La hipermocistinemia, independientemente de la causa, provoca ateroesclerosis, quizá por varios efecto directos e indirectos sobre el endotelio vascular.100 Es raro que los efectos enzimáticos causen elevaciones severas de homocisteína y daño vascular temprano, pero son comunes las elevaciones moderadas, que quizá deriven de causas dietéticas y genéticas. Varios estudios observacionales han encontrado una asociación positiva entre la concentración de homocisteína y la ateroesclerosis,101,102 pero no existen estudios clínicos aleatorios que analicen el efecto de disminuir los niveles de homocisteína. La concentración de homocisteína en ayuno mayor de 15 µmol/L se asocia con un incremento suficiente en el riesgo en los pacientes con enfermedad vascular como para considerar el tratamiento. Los suplementos de folato, 1 a 5 mg por día, son seguros y casi siempre suficientes para disminuir la concentración de homocisteína a un rango adecuado. No es posible hacer recomendaciones firmes sobre el escrutinio o el tratamiento de las concentraciones elevadas de Lp(a) u homocisteína por ausencia de datos obtenidos en estudios clínicos.
Tratamiento farmacológico de los trastornos de las lipoproteínas
La elección de los medicamentos para los diferentes trastornos de los lípidos debe guiarse por la concentración de triglicéridos [ver tabla 6]. El médico debe también considerar aspectos como los efectos adversos, el costo y la conveniencia [ver tabla 7].
|
||||||||||||
* Si la LAD es baja, es preferible la niacina.
|
|
|||
LBD--lipoproteínas de baja densidad LAD--lipoproteínas de alta densidad LMBD--lipoproteínas de muy baja densidad reductasa de HMG-CoA--reductasa de la 3-hidroxi-3-metilglutaril coenzima A CK--creatina cinasa |
FARMACOS PARA EL AUMENTO EN LA CONCENTRACION DE COLESTEROL DE LBD
Para el tratamiento de las alteraciones de lípidos en donde el problema principal es el aumento en la concentración de colesterol de LBD (con triglicéridos normales o casi normales), las resinas que fijan ácidos biliares son los medicamentos de elección por su baja toxicidad. Estos fármacos son bastante seguros, pero están disponibles sólo en forma de polvo y deben ser hidratados antes de usarse, o ingerirse en tabletas de dosis baja, lo que los hace incómodos. Muchos pacientes los toleran sin efecto colaterales, lo que aumenta su constancia, siempre y cuando el médico los aliente e indique algunos consejos claves para facilitar el uso del medicamento. Estos consejos incluyen agregar una preparación de psyllium a la resina si ocurre constipación y preparar una dosis de resina y agua (o jugo) para tres días y mantenerla en el refrigerador. Las resinas pueden ser eficaces en dosis bajas,103 y se dispone de tabletas de colestipol de 1g, que muchos pacientes prefieren si toman 12 g o menos al día.
Las estatinas son también medicamentos de primera elección para disminuir el nivel de colesterol de LBD. Los autores aún prefieren las resinas en pacientes jóvenes y con riesgo limítrofe, pero las estatinas son muy eficaces y seguras,19 además de cómodas y relativamente libres de efectos adversos. Las cinco estatinas disponibles son equivalentes respecto a uso, aunque difieren un poco en potencia [ver tabla 7]. El precio de estos agentes está determinado por contratos entre los provedores y compradores como los HMO, que tienen tanto costos reducidos como menor nivel de flexibilidad para prescribir medicamentos. La combinación de una resina y una estatina es muy eficaz porque aumenta el número de receptores hepáticos de LBD por mecanismos complementarios.
El ácido nicotínico es también eficaz para disminuir el colesterol de LBD, reduciendo la concentración de LMBD y aumentando las LAD. Los autores lo utilizan como medicamento único o en combinación con resinas en pacientes con hiperlipidemia mixta en quienes el uso aislado de resinas puede aumentar la concentración de triglicéridos. El ácido nicotínico tiene muchos efectos colaterales, incluyendo la elevación de las enzimas hepáticas, que deben ser vigiladas en forma periódica. Sin embargo, puede ser eficaz en dosis bajas (1.0 a 1.5 g/día), que muchos pacientes toleran.
Aunque el gemfibrozil aumenta las LAD y disminuye las LMBD y los triglicéridos, es menos eficaz que otros fármacos para disminuir las LBD. Sin embargo, es bien tolerado y puede ser usado en pacientes seleccionados con hiperlipidemia mixta. Los individuos que participaron en el Estudio del Corazón de Helsinki y que se beneficiaron más por el tratamiento con gemfibrozil para la prevención primaria de la enfermedad coronaria, tuvieron una relación de LBD a LAD mayor de 5 y una concentración de triglicéridos por arriba de 200 mg/dl.47 Sin embargo, existe preocupación sobre la seguridad de los fibratos.93
FARMACOS USADOS PRINCIPALMENTE PARA EL AUMENTO EN LOS TRIGLICERIDOS
El gemfibrozil es el medicamento mejor tolerado cuando se trata el aumento de triglicéridos y LMBD en ausencia de incrementos significativos en el colesterol de LBD. Como el colesterol de LAD es bajo en estos pacientes, el efecto del gemfibrozil sobre las LAD es también benéfico. Algunos pacientes experimentan un aumento en el colesterol del LBD con el uso de gemfibrozil (casi siempre cuando las LBD están disminuidas), este efecto parece ser resultado de un mayor catabolismo de las LMBD, que causa aumento en la producción de las LBD. Si las LBD exceden 130 mg/dl, deberá iniciarse tratamiento para disminuirlas. El clofibrato, que es el ácido fíbrico original, no se utiliza ya porque se asoció con un gran riesgo no cardiovascular en un estudio clínico. Sin embargo, puede administrarse en algunos pacientes con aumento severo de triglicéridos y que no toleran gemfibrozil o ácido nicotínico. Siempre es importante destacar que el ejercicio y la pérdida de peso son indispensables para controlar el aumento en los triglicéridos, y que nunca deben abandonarse los esfuerzos por modificar estos patrones de vida.
REMPLAZO ESTROGENICO EN MUJERES POSMENOPAUSICAS
Los estudios epidemiológicos muestran que la incidencia de infarto al miocardio en las mujeres posmenopáusicas que reciben tratamiento sustitutivo con estrógenos es alrededor de la mitad que en las pacientes que no reciben este tratamiento. El estrógeno aumenta la LAD, disminuye la LBD, probablemente disminuye la Lp(a) y el fibrinógeno, e inhibe la oxidación de LBD. Los estrógenos aumentan la producción de triglicéricos, pero no disminuyen su depuración, y no se piensa que esta propiedad sea aterogénica. La adición de una progestina reduce la magnitud del incremento en la concentración de LAD causada por los estrógenos sin oposición, pero también reduce el riesgo de hiperplasia endometrial adenomatosa o atípica en mujeres con útero.83 El tratamiento con estrógenos puede aumentar el riesgo de cáncer de mama, este riesgo parece ser pequeño, pero aumenta con el uso prolongado.104 Si el beneficio vascular del tratamiento estrogénico alcanza al observado en estudios epidemiológicos, la reducción en el riesgo de enfermedad cardiovascular supera el mayor riesgo de cáncer. En la actualidad se realizan estudios extensos de prevención primaria y secundaria para valorar la eficacia del tratamiento estrogénico en la reducción de eventos coronarios. Hasta que estén listos los resultados, es necesario individualizar la necesidad de tratamiento, tomando en cuenta otras indicaciones para sustitución estrogénica, como osteoporosis y síntomas posmenopáusicos, grado de riesgo de enfermedad coronaria y riesgo potencial del tratamiento estrogénico prolongado. Los lineamientos del NCEP recomiendan el tratamiento estrogénico sustitutivo como una alternativa a los medicamentos hipolipemiantes para mujeres posmenopáusicas que requieren tratamiento farmacológico para disminuir la LBD, pero las evidencias de estudios clínicos favorecen el uso de estatinas, por lo menos en las mujeres con enfermedad coronaria.
ACIDOS GRASOS OMEGA-3 (ACEITE DE PESCADO)
El interés en los aceites de pescado comenzó con la observación de que los esquimales de Groenlandia, que ingieren una dieta rica en grasa (40 porciento de sus calorías totales), tienen baja incidencia de enfermedad cardiaca. La grasa que existe en el pescado es rica en ácidos grasos no saturados omega-3. El efecto principal de los ácidos grasos omega-3 sobre los lípidos consiste en disminuir los triglicéridos, aunque el fecto sobre la LBD es variable. No se sabe con exactitud qué dosis se requiere para causar modificaciones en los lípidos, en algunos estudios se han utilizado dosis entre 15 a 30 g/día, mientras que en otros se indica que dosis de 2 g/día o menos son eficaces.
En la actualidad existen muchos suplementos de aceite de pescado en el mercado que, por no ser considerados como medicamentos, no están controlados por la FDA. Aún se desconoce si es seguro ingerir cápsulas de aceite de pescado por largo plazo, y no existen evidencias de que los suplementos de aceite de pescado eviten la enfermedad cardiaca.
Aspectos especiales del tratamiento de las alteraciones de los lípidos
TRATAMIENTO DE LA HIPERCOLESTEROLEMIA EN MUJERES
Las mujeres tienen un menor riesgo general de enfermedad cardiovascular que los hombres, y esto ha hecho que algunos médicos consideren que es inapropiado investigar en ellas las concentraciones de colesterol.105 De hecho, la enfermedad cardiovascular es la primera causa de muerte tanto en hombres como en mujeres, sólo que estas últimas mueren por enfermedad cardiovascular 10 años después de que los varones. Esto significa que las mujeres en la séptima década de la vida tienen la misma mortalidad cardiovascular que los hombres en la sexta. Es más, el valor del escrutinio periódico del colesterol es sobre todo educacional, permitiendo a quienes tienen alto riesgo beneficiarse de la información disponible sobre cómo disminuir este riesgo. El objetivo médico de los estudios de escrutinio (encontrar casos) es secundario, porque la mayoría de la gente con una concentración de colesterol por arriba de 200 mg/dl requiere sólo de modificaciones en la dieta y otros cambios en el estilo de vida, y no tratamiento médico. Sin embargo, el riesgo inmediato y absoluto para las mujeres con una elevación determinada en el colesterol de LBD es menor que para los hombres con el mismo aumento, y las intervenciones deben depender del grado de riesgo.82 Este objetivo se toma en cuenta en parte en las normas del NCEP, que combinan la edad y el sexo como un factor de riesgo. Los autores también utilizan los fármacos que disminuyen el riesgo (como las resinas) cuando es necesario, y establecen cifras ideales de LBD (quizá 150 en lugar de 130 mg/dl), en especial si la concentración de colesterol de LAD es alta. De hecho, las LAD pueden ser más importantes en las mujeres que en los hombres,84 y es poco probable que las mujeres con colesterol de LAD por arriba de 60 mg/dl requieran disminuir sus LBD a menos de que su concentración sea mayor a 220 mg/dl. Los estudios de prevención secundaria han encontrado un beneficio semejante tanto en mujeres como en varones, de modo que es apropiada la reducción agresiva del riesgo en ambos sexos.19,20
ESCRUTINIO Y TRATAMIENTO DE LA HIPERCOLESTEROLEMIA EN NIÑOS Y ADULTOS JOVENES
Los médicos que atienden a adultos con enfermedad coronaria temprana o alguna alteración de lípidos deben enfrentarse a la decisión de si debe investigarse y tratarse la hipercolesterolemia en los niños. Numerosos estudios de necropsia demuestran que la ateroesclerosis coronaria comienza en la infancia y adolescencia, y que las concentraciones de lipoproteínas se asocian en forma consistente en el grado de esta ateroesclerosis. Los niños de familias con alteraciones de lípidos o enfermedad coronaria temprana tienen concentraciones más altas de colesterol, y los niveles de colesterol durante la infancia predicen los niveles que existirán en la edad adulta. Sin embargo, una proporción importante de niños y adolescentes con colesterol elevado no tendrán, en la edad adulta, concentraciones tan altas como para justificar su tratamiento, y el escrutinio del nivel de colesterol en todos los niños podría causar efectos negativos al etiquetar a mucha gente joven como enferma. Todos los niños pueden beneficiarse de una dieta pobre en grasas saturadas, y esta medida debe ser parte de la estrategia de cualquier población para controlar la ateroesclerosis epidémica. Sin embargo, la seguridad y eficacia del tratamiento farmacológico a largo plazo en este grupo de edad no han sido establecidos, y el tratamiento debe administrarse en forma muy cuidadosa.
Considerando éste y otros aspectos, los autores están de acuerdo con las recomendaciones del National Cholesterol Education Programs Expert Panel on Blood Cholesterol Levels in Children and Adolescents (Mesa redonda de especialidades del programa nacional de educación sobre colesterol acerca de las concentraciones plasmáticas de colesterol en niños y adultos, n del t.).106 Los médicos deben aconsejar a los pacientes menores de 55 años con enfermedad coronaria diagnosticada o alguna alteración de lípidos para que se mida en forma periódica el colesterol en sus hijos o nietos en una institución de atención continua. Por supuesto, los pacientes con alguno de los raros padecimientos genéticos del metabolismo de lípidos deben solicitar consejo genético apropiado. Los médicos que atiendan a un paciente joven (i.e., menor de 20 años) con aumento marcado en la concentración de LBD deben agotar todos los cambios sobre el estilo de vida antes de considerar la administración de fármacos. Si estas medidas son ineficaces, las resinas son el medicamento de elección, y deberá considerarse referir al paciente a una clínica especializada.
El tratamiento de los adultos jóvenes con colesterol elevado también es tema de controversia.105 Los autores están de acuerdo en la estrategia de equiparar la intervención al nivel de riesgo,82 pero el riesgo a corto plazo (v.gr., 10 años) en adultos jóvenes puede proporcionar un cálculo inadecuado sobre el beneficio potencial de reducir el colesterol. Law, Wald y Thompson83 mostraron que los diferentes tipos de estudios indican que el mayor beneficio de la reducción de colesterol se logra en personas jóvenes, con una reducción del riesgo durante la vida hasta del 50 porciento a los 40 años si se reduce el colesterol en un 10 porciento. Además, la muerte súbita sigue siendo una manifestación inicial importante de enfermedad coronaria, y la tasa de enfermedad coronaria en los grupos de tratamiento activo en los estudios clínicos sigue siendo demasiado alta. Por lo tanto, es incorrecto afirmar que el tratamiento puede diferirse con seguridad hasta una fase tardía de la vida. Deben fomentarse en los jóvenes las medidas de prevención e intervención relativas al estilo de vida, pero se requiere de los avances de la tecnología para identificar mejor a los pacientes asintomáticos (de cualquier edad) que deben someterse a reducción agresiva de los factores de riesgo. Estos adelantos permitirán identificar o cuantificar placas vulnerables, marcadores de inflamación o medidas no invasivas de disfunción endotelial.
TRATAMIENTO DE LA HIPERCOLESTEROLEMIA EN EL ANCIANO
En la actualidad está bien establecido que la concentración de colesterol predice el riesgo de ateroesclerosis en las personas mayores de 65 años,107,108 y que la mayor parte de las enfermedades cardiovasculares ocurren a partir de esta edad, en especial en las mujeres. El Scandinavian Simvastatin Survival Study y el Cholesterol and Recurrent Events Trial encontró una reducción significativa en la mortalidad total y en los eventos coronarios importantes en individuos mayores de 60 años.19,20 Sin embargo, no se sabe si las medidas para disminuir el colesterol son eficaces en los ancianos. Los autores recomiendan individualizar en forma cuidadosa el tratamiento para estos pacientes. Los padecimientos concomitantes importantes contraindican hasta cierto punto el tratamiento preventivo, pero los ancianos sanos y vigorosos pueden recibir tratamiento para una dislipoproteinemia, usando las medidas de bajo riesgo tanto como sea posible y estableciendo metas moderadas. El consejo dietético debe ser individualizado para asegurar una nutrición adecuada en personas cuya alimentación puede ser marginal. La adición de cereal de avena o psyllium puede ser benéfica, pero las dosis deben iniciarse en forma gradual para evitar un impacto fecal. Las resinas también pueden ser útiles en casos seleccionados.
COLESTEROL BAJO Y MORTALIDAD NO CARDIOVASCULAR
La relación entre el colesterol bajo y la salud ha sido tema de controversia desde hace mucho tiempo.109 Aunque en la actualidad se acepta en forma generalizada que existe una relación causa-efecto entre las dietas con grasas saturadas y el colesterol plasmático, y entre el colesterol plasmático y la enfermedad ateroesclerótica, siguen existiendo dudas en relación con el posible aumento de enfermedades no cardiovasculares en los individuos con concentraciones bajas de colesterol en plasma. Esta controversia deriva tanto de estudios observacionales110 como de estudios de prevención primaria111, y ha provocado que algunos médicos sugieran que no debe tratarse a los pacientes con concentraciones de colesterol elevado.112 Existen publicaciones detalladas al respecto.113
Es posible que dos fenómenos expliquen la conexión entre las concentraciones bajas de colesterol y las muertes no cardiovasculares: la inversión de la causa y la confusión. La inversión de la causa (i.e., el efecto se aprecia como causa) se refiere en este caso al hecho de que enfermedades previas puedan disminuir la concentración de colesterol. Aunque las muertes tempranas se eliminaron de estos estudios de cohorte para compensar este efecto, la disminución en la concentración de colesterol pudo ocurrir más de 10 años antes del diagnóstico.114 Debido a que muchas enfermedades diferentes se asocian con concentraciones bajas de colesterol, este dato puede ser solo un indicador de mala salud y nutrición. Esta afirmación es apoyada por los datos de seguimiento a largo plazo,115 estudios realizados en China,116 un estudio que consideró la clase social,117 y los análisis recientes del Estudio del corazón realizado en Hawaii.118
La mayoría de los datos al respecto provienen de estudios de prevención primaria de reducción del colesterol, que no han demostrado una mortalidad total menor en los grupos tratados debido a frecuencias mayores de muertes por causas no cardiovasculares.109 Los primeros meta-análisis de estos estudios no fueron adecuados porque combinaban estudios que no eran semejantes: algunos consistían en intervenciones dietéticas, mientras que otros eran farmacológicos y comparaban diferentes agentes. Además, algunos mostraban un exceso en la mortalidad por cáncer y en otros existían causas de muerte diferentes. El único hallazgo constante en los estudios es un aumento en las muertes por violencia. Sin embargo, el número de muertes violentas es pequeño (dos de 21 en el grupo tratado y cero de 15 en el grupo control). Un meta-análisis de 1995 de estudios sobre agentes hipolipemiantes ha ayudado a aclarar este aspecto.93 Este meta-análisis es diferente de varios realizados antes en tres aspectos principales: es el meta-análisis más completo, examinó la heterogeneidad de efectos en las diferentes intervenciones y analizó la relación dosis respuesta entre la reducción de colesterol y todas las evoluciones (un efecto gradual, positivo o negativo, reforza la inferencia causal). Este meta-análisis encontró que por cada 10 porciento de puntos de reducción del colesterol, la mortalidad por enfermedad coronaria disminuyó en 13 porciento (P< 0.002) y que la mortalidad total se redujo en 10 porciento (P< 0.03). Es importante mencionar que el análisis mostró también que los fibratos (clofibrato, en siete estudios y gemfibrozil, en dos) aumentaron la mortalidad por enfermedad no coronaria en alrededor del 30 porciento (P< 0.01) y la mortalidad total en alrededor del 17 porciento (P < 0.02). El aspecto del riesgo no cardiovascular por la disminución del colesterol ha dejado de preocupar por los resultados de los estdios de prevención tanto primaria como secundaria con estatinas, que producen reducciones mayores de colesterol que los estudios previos y que han demostrado que no existe aumento en las muertes no cardiovasculares y sí una reducción significativa en la mortalidad global.16,19,20
El médico debe saber que la prevención primaria se practica en pacientes asintomáticos, lo que requiere evaluar la relación costo-beneficio en ausencia de la necesidad de aliviar síntomas. No es fácil extrapolar, a partir de los estudios clínicos, los costos y beneficios de un tratamiento en especial para un paciente específico. Por lo tanto, está indicado tener precaución, sobre todo cuando se prescriben medicamentos por tiempo prolongado. Los cambios en la nutrición y el ejercicio deben ser las medidas principales de intervención primaria para la enfermedad cardiovascular tanto en forma individual como para la población en general. Los pacientes con enfermedad coronaria establecida tienen riesgo muy alto de eventos subsecuentes, y definitivamente se benefician con la reducción de lípidos, ameritando control agresivo de la LBD, con frecuencia por medio de fármacos.
Figuras 1 a 4 Andy Christie.
Bibliografía
- Fredrickson DS, Levy RI, Lees RS: Fat transport and lipoproteins-an integrated approach to mechanisms and disorders. N Engl J Med 276:32, 94, 148, 215, 273, 1967
- Stamler J, Wentworth D, Neaton JD: Is relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Findings in 356,222 primary screenees of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA 256:2823, 1986 [PMID 3773199]
- Young SG: Recent progress in understanding apolipoprotein B. Circulation 82:1574, 1990
- Brown MS, Goldstein JL: A receptor-mediated pathway for cholesterol homeostasis. Science 232:34, 1986 [PMID 3513311]
- Eisenberg S: High density lipoprotein metabolism. J Lipid Res 25:1017, 1984
- Tall AR: Plasma high density lipoproteins: metabolism and relationship to atherogenesis. J Clin Invest 86:379, 1990
- Rothblat GH, Mahlberg FH, Johnson WJ, et al: Apolipoproteins, membrane cholesterol domains, and the regulation of cholesterol efflux. J Lipid Res 33:1091, 1992 [PMID 1431592]
- Rubin EM, Krauss RM, Spangler EA, et al: Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 353:265, 1991 [PMID 1910153]
- Warden CH, Hedrick CC, Qiao JH, et al: Atherosclerosis in transgenic mice overexpressing apolipoprotein A-II. Science 261:469, 1993 [PMID 8332912]
- Parthasarathy S, Barnett J, Fong L: High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim Biophys Acta 1044:275, 1990 [PMID 2344447]
- Scanu AM: Lipoprotein(a): a genetic risk factor for premature coronary heart disease. JAMA 267:3326, 1992
- Eaton SB, Konner M: Paleolithic nutrition: a consideration of its nature and current implications. N Engl J Med 312:283, 1985 [PMID 2981409]
- Blackburn H: Diet and mass hyperlipidemia: a public health view. Nutrition, lipids, and coronary heart disease. Levy R, Rifkind B, Dennis B, et al, Eds. Raven Press, New York, 1979, p 309
- Farquhar JW, Fortmann SP, Flora JA, et al: Effects of communitywide education on cardiovascular disease risk factors: the Stanford Five-City Project. JAMA 264:359, 1990
- The Lipid Research Clinics Coronary Primary Prevention Trial results: I. Reduction in incidence of coronary heart disease. Lipid Research Clinics Program. JAMA 251:351, 1984
- Shepherd J, Cobbe SM, Ford I, et al: Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med 333:1301, 1995 [PMID 7566020]
- Frick MH, Elo O, Haapa K, et al: Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. N Engl J Med 317:1237, 1987 [PMID 3313041]
- Brown BG, Zhao X-Q, Sacco DE, et al: Lipid lowering and plaque regression: new insights into prevention of plaque disruption and clinical events in coronary disease. Circulation 87:1781, 1993 [PMID 8504494]
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Scandinavian Simvastatin Survival Study Group. Lancet 344:1383, 1994
- Sacks FM, Pfeffer MA, Moye LA, et al: The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med 335:1001, 1996
- Fuster V, Badimon L, Badimon JJ, et al: The pathogenesis of coronary artery disease and the acute coronary syndromes (pt 2). N Engl J Med 326:310, 1992 [PMID 1728735]
- Levine GN, Keaney JF Jr, Vita JA: Cholesterol reduction in cardiovascular disease: clinical benefits and possible mechanisms. N Engl J Med 332:512, 1995 [PMID 7830734]
- Flavahan NA: Atherosclerosis or lipoprotein-induced endothelial dysfunction: potential mechanisms underlying reduction in EDRF/nitric oxide activity. Circulation 85:1927, 1992
- Zeiher AM, Drexler H, Wollschlager H, et al: Modulation of coronary vasomotor tone in humans: progressive endothelial dysfunction with different early stages of coronary atherosclerosis. Circulation 83:391, 1991 [PMID 1991363]
- Treasure CB, Klein JL, Weintraub WS, et al: Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med 332:481, 1995 [PMID 7830728]
- Anderson TJ, Meredith IT, Yeung AC, et al: The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med 332:488, 1995 [PMID 7830729]
- van Boven AJ, Jukema JW, Zwinderman AH, et al: Reduction of transient myocardial ischemia with pravastatin in addition to the conventional treatment in patients with angina pectoris. Circulation 94:1503, 1996 [PMID 8840836]
- Andrews TC, Raby K, Barry J, et al: Effect of cholesterol reduction on myocardial ischemia in patients with coronary disease. Circulation 95:324, 1997 [PMID 9008444]
- Summary of the second report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II). Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. JAMA 269:3015, 1993
- Steinberg D, Parthasarathy S, Carew TE, et al: Beyond cholesterol: modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med 320:915, 1989
- Stephens NG, Parsons A, Schofield PM, et al: Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 347:781, 1996 [PMID 8622332]
- Austin MA, Breslow JL, Hennekens CH, et al: Low-density lipoprotein subclass patterns and risk of myocardial infarction. JAMA 260:1917, 1988 [PMID 3418853]
- Austin MA, Brunzell JD, Fitch WL, et al: Inheritance of low density lipoprotein subclass patterns in familial combined hyperlipidemia. Arteriosclerosis 10: 520, 1990 [PMID 2369363]
- Gardner CD, Fortmann SP, Krauss RM: Association of small low-density lipoprotein particles with the incidence of coronary artery disease in men and women. JAMA 276:875, 1996 [PMID 8782636]
- Stampfer MJ, Sacks FM, Salvini S, et al: A prospective study of cholesterol, apolipoproteins, and the risk of myocardial infarction. N Engl J Med 325:373, 1991 [PMID 2062328]
- McMurry MP, Cerqueira MT, Connor SL, et al: Changes in lipid and lipoprotein levels and body weight in Tarahumara Indians after consumption of an affluent diet. N Engl J Med 325:1704, 1991 [PMID 1944471]
- Badimon JJ, Badimon L, Galvez A, et al: High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab Invest 60:455, 1989 [PMID 2927083]
- Badimon JJ, Badimon L, Fuster V: Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest 85:1234, 1990 [PMID 2318976]
- Genest J Jr, McNamara JR, Ordovas JM, et al: Lipoprotein cholesterol, apolipoprotein A-I and B and lipoprotein (a) abnormalities in men with premature coronary artery disease. J Am Coll Cardiol 19:792, 1992 [PMID 1531990]
- Miller M, Seidler A, Kwiterovich PO, et al: Long-term predictors of subsequent cardiovascular events with coronary artery disease and "Ldesirable"R levels of plasma total cholesterol. Circulation 86:1165, 1992 [PMID 1394924]
- Levy RI, Brensike JF, Epstein SE, et al: The influence of changes in lipid values induced by cholestyramine and diet on progression of coronary artery disease: results of the NHLBI Type II Coronary Intervention Study. Circulation 69:325, 1984 [PMID 6360415]
- Brown G, Albers JJ, Fisher LD, et al: Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med 323:1289, 1990 [PMID 2215615]
- Smith SC Jr, Blair SN, Criqui MH, et al: Preventing heart attack and death in patients with coronary disease. American Heart Association Consensus Panel Statement. Circulation 92:2, 1995
- Baseline serum cholesterol and treatment effect in the Scandinavian Simvastatin Survival Study (4S). Scandinavian Simvastatin Survival Study Group. Lancet 345:1274, 1995
- Criqui MH, Heiss G, Cohn R, et al: Plasma triglyceride level and mortality from coronary heart disease. N Engl J Med 328:1220, 1993 [PMID 8464432]
- Assmann G, Schulte H: Triglycerides and atherosclerosis: results from the Prospective Cardiovascular Münster Study. Atherosclerosis Reviews 22:51, 1991
- Manninen V, Tenkanen L, Koskinen P, et al: Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study: implications for treatment. Circulation 85:37, 1992 [PMID 1728471]
- Carlson LA, Rosenhamer G: Reduction of mortality in the Stockholm Ischaemic Heart Disease Secondary Prevention Study by combined treatment with clofibrate and nicotinic acid. Acta Med Scand 223:405, 1988 [PMID 3287837]
- Castelli WP: Epidemiology of triglycerides: a view from Framingham. Am J Cardiol 70:3H, 1992
- Zilversmit DB: Atherogenesis: a postprandial phenomenon. Circulation 60:473, 1979
- Wild SH, Fortmann SP, Marcovina SM: A prospective case-control study of lipoprotein(a) levels and apo(a) size and risk of coronary heart disease in Stanford Five-City Project participants. Arterioscler Thromb Vasc Biol 17:239, 1997 [PMID 9081676]
- Seed M, Hoppichler F, Reaveley D, et al: Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia. N Engl J Med 322:1494, 1990 [PMID 2139920]
- Bostom AG, Gagnon DR, Cupples LA, et al: A prospective investigation of elevated lipoprotein (a) detected by electrophoresis and cardiovascular disease in women: the Framingham Heart Study. Circulation 90:1688, 1994 [PMID 7923652]
- Schaefer EJ, Lamon-Fava S, Jenner JL, et al: Lipoprotein(a) levels and risk of coronary heart disease in men: the Lipid Research Clinics Coronary Primary Prevention Trial. JAMA 271:999, 1994 [PMID 8139085]
- Ridker PM, Hennekens CH, Stampfer MJ: A prospective study of lipoprotein(a) and the risk of myocardial infarction. JAMA 270:2195, 1993 [PMID 8411602]
- Ridker PM, Stampfer MJ, Hennekens CH: Plasma concentration of lipoprotein(a) and the risk of future stroke. JAMA 273:1269, 1995 [PMID 7715039]
- Fortmann SP, Marcovina SM: Lipoprotein(a), a clinically elusive lipoprotein particle (editorial). Circulation 95:295, 1997 [PMID 9008434]
- Consensus conference: Treatment of hypertriglyceridemia. JAMA 251:1196, 1984
- Santamarina-Fojo S, Brewer HB Jr: The familial hyperchylomicronemia syndrome: new insights into the underlying genetic defects. JAMA 265:904, 1991 [PMID 1992190]
- Schonfeld G: Inherited disorders of lipid transport. Endocrinol Metab Clin North Am 19:229, 1990
- Benlian P, De Gennes JL, Foubert L, et al: Premature atherosclerosis in patients with familial chylomicronemia caused by mutations in the lipoprotein lipase gene. N Engl J Med 335:848, 1996 [PMID 8778602]
- Goldstein JL, Schrott HG, Hazzard WR, et al: Hyperlipidemia in coronary heart disease: II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest 52:1544, 1973
- Sanderson S, Iverius PH, Wilson DE: Successful hyperlipemic pregnancy. JAMA 265:1858, 1991
- Ginsberg HN: Lipoprotein physiology and its relationship to atherogenesis. Endocrinol Metab Clin North Am 19:211, 1990
- Kane JP, Havel RJ: Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. The Metabolic Basis of Inherited Disease, 6th ed. Scriver CR, Beaudet AL, Sly WS, et al, Eds. McGraw Hill Book Co, New York, 1989, p 1139
- Hobbs HH, Russell DW, Brown MS, et al: The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet 24:133, 1990 [PMID 2088165]
- Torsvik H, Fischer JE, Feldman HA, et al: Effects of intravenous hyperalimentation on plasma-lipoproteins in severe familial hypercholesterolaemia. Lancet 1:601, 1975 [PMID 47947]
- Bilheimer DW, Goldstein JL, Grundy SM, et al: Liver transplantation to provide low-density-lipoprotein receptors and lower plasma cholesterol in a child with homozygous familial hypercholesterolemia. N Engl J Med 311:1658, 1984 [PMID 6390206]
- Kane JP, Malloy MJ, Ports TA, et al: Regression of coronary atherosclerosis during treatment of familial hypercholesterolemia with combined drug regimens. JAMA 264: 3007, 1990 [PMID 2243428]
- Illingworth DR, Vakar F, Mahley RW, et al: Hypocholesterolaemic effects of lovastatin in familial defective apolipoprotein B-100. Lancet 339:598, 1992 [PMID 1347103]
- Mahley RW, Weisgraber KH, Innerarity TL, et al: Genetic defects in lipoprotein metabolism: elevation of atherogenic lipoproteins caused by impaired catabolism. JAMA 265:78, 1991 [PMID 1845776]
- Forte TM, Carlson LA: Electron microscopic structure of serum lipoproteins from patients with fish eye disease. Arteriosclerosis 4:130, 1984 [PMID 6704050]
- Franceschini G, Sirtori CR, Capurso A, et al: A-IMilano apoprotein: decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest 66:892, 1980
- Franceschini G, Vecchio G, Gianfranceschi G, et al: Apolipoprotein AIMilano: accelerated binding and dissociation from lipids of a human apolipoprotein variant. J Biol Chem 260:16321, 1985 [PMID 3934174]
- Norum RA, Lakier JB, Goldstein S, et al: Familial deficiency of apolipoproteins A-I and C-III and precocious coronary-artery disease. N Engl J Med 306: 1513, 1982 [PMID 7078608]
- Garg A, Grundy SM: Nicotinic acid as therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. JAMA 264:723, 1990 [PMID 2374275]
- Chait A, Brunzell JD: Acquired hyperlipidemia (secondary dyslipoproteinemias). Endocrinol Metab Clin North Am 19:259, 1990 [PMID 2192873]
- Joven J, Villabona C, Vilella E, et al: Abnormalities of lipoprotein metabolism in patients with the nephrotic syndrome. N Engl J Med 323:579, 1990 [PMID 2381443]
- Steinberg D, Pearson TA, Kuller LH: Alcohol and atherosclerosis. Ann Intern Med 114:967, 1991
- Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women: the Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. The Writing Group for the PEPI Trial. JAMA 273:199, 1995
- Hulley SB, Newman TB, Grady D, et al: Should we be measuring blood cholesterol levels in young adults? JAMA 269:1416, 1993
- Fuster V, Gotto AM, Libby P, et al: Matching the intensity of risk factor management with the hazard for coronary disease events. Task Force 1. Pathogenesis of coronary disease: the biologic role of risk factors. 27th Bethesda Conference. J Am Coll Cardiol 27:957, 1996
- Law MR, Wald NJ, Thompson SG: By how much and how quickly does reduction in serum cholesterol concentration lower risk of ischaemic heart disease? BMJ 308:367, 1994
- Abbot RD, Wilson PWF, Kannel WB, et al: High density lipoprotein cholesterol, total cholesterol screening, and myocardial infarction: the Framingham Study. Arteriosclerosis 8:207, 1988
- Wood PD, Stefanick ML, Williams PT, et al: The effects on plasma lipoproteins of a prudent weight-reducing diet, with or without exercise, in overweight men and women. N Engl J Med 325:461, 1991 [PMID 1852180]
- Blum CD, Levy RI, Eisenberg S, et al: High density lipoprotein metabolism in man. J Clin Invest 60:795, 1977
- Sacks FM, Willett WW: More on chewing the fat: the good fat and the good cholesterol. N Engl J Med 325:1740, 1991 [PMID 1944476]
- Ripsin CM, Keenan JM, Jacobs DR, et al: Oat products and lipid lowering: a meta-analysis. JAMA 267:3317, 1992 [PMID 1317928]
- Bell LP, Hectorne K, Reynolds H, et al: Cholesterol-lowering effects of psyllium hydrophilic mucilloid: adjunct therapy to a prudent diet for patients with mild to moderate hypercholesterolemia. JAMA 261:3419, 1989 [PMID 2724486]
- Zavaroni I, Bonora E, Pagliara M, et al: Risk factors for coronary artery disease in healthy persons with hyperinsulinemia and normal glucose tolerance. N Engl J Med 320:702, 1989
- Wood PD, Stefanick ML, Dreon DM, et al: Changes in plasma lipids and lipoproteins in overweight men during weight loss through dieting as compared with exercise. N Engl J Med 319:1173, 1988 [PMID 3173455]
- Superko HR, Krauss RS: Differential effects of nicotinic acid in subjects with different LDL subclass patterns. Atherosclerosis 95:69, 1992 [PMID 1642694]
- Gould AL, Rossouw JE, Santanello NC, et al: Cholesterol reduction yields clinical benefit: a new look at old data. Circulation 91:2274, 1995 [PMID 7697857]
- Kreisberg RA: Low high-density lipoprotein cholesterol: what does it mean, what can we do about it, and what should we do about it? Am J Med 94:1, 1993
- Kashyap ML: Effects of drugs on high-density lipoprotein apoprotein metabolism. High-Density Lipoproteins, Reverse Cholesterol Transport, and Coronary Heart Disease. Miller NE, Ed. Excerpta Medica, New York, 1989, p 60
- Shepherd J, Packard CJ, Morgan HG, et al: The effects of cholestyramine on high density lipoprotein metabolism. Atherosclerosis 33:433, 1979 [PMID 228682]
- Saku K, Gartside PS, Hynd BA, et al: Mechanism of action of gemfibrozil on lipoprotein metabolism. J Clin Invest 75:1702, 1985 [PMID 3923042]
- Shepherd J, Packard CJ, Patsch JR, et al: Effects of nicotinic acid therapy on plasma high density lipoprotein subfraction distribution and composition and on apolipoprotein A metabolism. J Clin Invest 63:858, 1979 [PMID 221531]
- Rubenfire M, Maciejko JJ, Blevins RD, et al: The effect of pravastatin on plasma lipoprotein and apolipoprotein levels in primary hypercholesterolemia. Arch Intern Med 151:2234, 1991
- Mayer EL, Jacobsen DW, Robinson K: Homocysteine and coronary atherosclerosis. J Am Coll Cardiol 27:517, 1996 [PMID 8606260]
- Graham IM, Daly LE, Refsum HM, et al: Plasma homocysteine as a risk factor for vascular disease. The European Concerted Action Project. JAMA 277:1775, 1997
- Nygard O, Nordrehaug JE, Refsum H, et al: Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med 337: 230, 1997 [PMID 9227928]
- Denke MA, Grundy SM: Efficacy of low-dose cholesterol-lowering drug therapy in men with moderate hypercholesterolemia. Arch Intern Med 155:393, 1995 [PMID 7848022]
- Grodstein F, Stampfer MJ, Colditz GA, et al: Postmenopausal hormone therapy and mortality. N Engl J Med 336:1769, 1997 [PMID 9187066]
- Guidelines for using serum cholesterol, high-density lipoprotein cholesterol, and triglyceride levels as screening tests for preventing coronary heart disease in adults. Clinical guideline, pt. 1. American College of Physicians. Ann Intern Med 124:515, 1996
- Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. American Academy of Pediatrics. National Cholesterol Education Program. Pediatrics 89:525, 1992
- Benfante R, Reed D: Is elevated serum cholesterol level a risk factor for coronary heart disease in the elderly? JAMA 263:393, 1990
- Shipley MJ, Pocock SJ, Marmot MG: Does plasma cholesterol concentration predict mortality from coronary heart disease in elderly people? 18 year follow up in Whitehall Study. BMJ 303:89, 1991 [PMID 1860009]
- Mann GV: Diet-heart: end of an era. N Engl J Med 297:644, 1977
- Jacobs D, Blackburn H, Higgins M, et al: Report of the conference on low blood cholesterol: mortality associations. Circulation 86:1046, 1992 [PMID 1355411]
- Muldoon MF, Manuck SB, Matthews KA: Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ 301:309, 1990 [PMID 2144195]
- Hulley SB, Walsh JMB, Newman TB: Health policy on blood cholesterol: time to change directions. Circulation 86:1026, 1992 [PMID 1516172]
- Epstein FH: Low serum cholesterol, cancer and other noncardiovascular disorders. Atherosclerosis 94:1, 1992
- Winawer SJ, Flehinger BJ, Buchalter J, et al: Declining serum cholesterol levels prior to diagnosis of colon cancer: a time-trend, case-control study. JAMA 263: 2083, 1990 [PMID 2319669]
- Anderson KM, Castelli WP, Levy D: Cholesterol and mortality: 30 years of follow-up from the Framingham Study. JAMA 257:2176, 1987 [PMID 3560398]
- Chen Z, Peto R, Collins R, et al: Serum cholesterol concentration and coronary heart disease in population with low cholesterol concentrations. BMJ 303:276, 1991 [PMID 1888927]
- Smith GD, Shipley MJ, Marmot MG, et al: Plasma cholesterol concentration and mortality: the Whitehall Study. JAMA 267:70, 1992 [PMID 1727199]
- Iribarren C, Dwyer JH, Burchfiel CM, et al: Can the U-shaped relation between mortality and serum cholesterol be explained by confounding? Circulation 87:684, 1993