Endocrinología
⭳ Abrir artículo (PDF)714.7 KBEste artículo es idéntico en la Edición 3/2000.
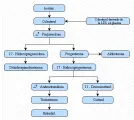
Contenido del artículo
IV SUPRARRENALES
- Fisiología de la corteza suprarrenal
- Hiperfunción de la corteza suprarrenal: Síndrome de Cushing
- EXCESO DE ACTH
- Producción hipofisaria excesiva (Enfermedad de Cushing)
- Producción ectópica de ACTH
- Producción ectópica de CRH
- EXCESO DE CORTISOL SUPRARRENAL
- DIAGNOSTICO
- TRATAMIENTO
- Síndrome de Nelson
- Uso farmacológico de los glucocorticoides
- Aldosteronismo primario
- Hipofunción de la corteza suprarrenal
- Síndromes de deficiencia poliendócrina
- Neoplasias suprarrenales e incidentalomas
- Hiperplasia suprarrenal congénita
- Hirsutismo
- La médula suprarrenal
IV SUPRARRENALES
DR. DANIEL D. FEDERMAN
Fisiología de la corteza suprarrenal
La glándula suprarrenal controla los ajustes del organismo en la posición erecta y permite la adaptación a la ingestión intermitente de alimentos. En ocasiones se requieren también aumentos súbitos y sostenidos en la función suprarrenal para tolerar episodios agudos de estrés como la pérdida de volumen, la infección, la anestesia y la cirugía. La pérdida de estas capacidades constituye la consecuencia más importante de la insuficiencia suprarrenal. Las secreciones suprarrenales también ejercen una influencia sobre la respuesta inmune, la formación de células sanguíneas, la función cerebral, la síntesis de proteínas y sobre muchos otros procesos que se llevan a cabo en el organismo.1 Al exceso de estos efectos se le conoce como síndrome de Cushing.
BlOSlNTESlS Y METABOLISMO DE LOS ESTEROIDES SUPRARRENALES
La síntesis de las hormonas suprarrenales se inicia a partirdel acetato o del colesterol [ver figura 1]. El colesterol derivado de las lipoproteínas de baja densidad (LDL) parece ser el sustrato para la biosíntesis del 80 porciento de los esteroides. La conversión del colesterol a D5- pregnenolona es el punto principal de regulación por las hormonas tróficas que controlan la función suprarrenal; tanto la corticotropina como la angiotensina estimulan este paso. Después de la síntesis de pregnenolona las vías metabólicas divergen hacia la formación de cada uno de los productos biológicamente activos de la suprarrenal.
Cortisol En los seres humanos el cortisol es el principal glucocorticoide. Influye sobre el apetito y el bienestar, tiene un papel primordial en el mantenimiento de los niveles de glucosa en la sangre al promover la gluconeogénesis hepática, y afecta en forma indirecta la frecuencia cardiaca y la función de bomba al controlar la síntesis de epinefrina en la médula suprarrenal. El aumento en la secreción de cortisol es una característica crucial en la respuesta fisiológica al estrés y a la enfermedad.
De 13 a 20 mg de cortisol son secretados cada día por las zonas fascicular y reticular; inmediatamente después de ser liberado por la glándula, el cortisol se une a la µ- globulina transcortina, o globulina fijadora de cortisol. Como en el caso de la tiroxina, la mayor parte del cortisol circulante se encuentra inactivo, unido a las proteínas; una pequeña proporción está libre y es biológicamente activo. La reducción, a nivel hepático, del doble enlace en el anillo A de la molécula inactiva al cortisol; después de varias biotransformaciones y conjugaciones con glucurónidos o sulfatos, los metabolitos del cortisol se excretan como 17 hidroxiesteroides (17-OHS) en la orina.
Andrógenos Los andrógenos se producen en la zona fascicular y reticular; su producción varía considerablemente en las distintas etapas de la vida. El feto elabora grandes cantidades de andrógenos suprarrenales, en especial de sulfato de dehidroepiandrosterona (DHEAS), mientras que el niño produce muy poco.2 La producción aumenta durante la pubertad y alcanza niveles máximos en los adultos jóvenes, que rápidamente comienzan a disminuir a niveles más bien bajos en las personas mayores de cincuenta años de edad. En cambio, la secreción de la hormona adrenocorticotrópica (ACTH), único mecanismo de control conocido de la biosíntesis de andrógenos suprarrenales, no muestra tales fluctuaciones relacionadas con la edad. La regulación de la producción de andrógenos suprarrenales no es muy clara: se han postulado factores tanto derivados de la hipófisis como de la suprarrenal, aunque no se han confirmado. Los andrógenos suprarrenales son relativamente poco potentes, pero sirven como precursores para la conversión hepática a testosterona [ver figura 2]. La hiperfunción de esta vía metabólica en las mujeres produce hirsutismo y, en ocaciones, virilización.
La androstenediona, otro andrógeno suprarrenal, puede ser transformado por el hígado y el tejido adiposo a estrona, un estrógeno potente. De hecho, está es la principal fuente de estrógenos en niños y mujeres posmenopáusicas. El otro andrógeno importante, la dehidroepinadrosterona (DHEA), junto con su conjugado DHEAS, constituye un enigma. La DHEA tiene una potencia androgénica mínima, y aunque se han publicado algunas acciones no adrogénicas, ninguna parece tener importancia fisiológica demostrada. En forma semejante, la DHEAS puede metabolizarse a testos terona o a estrógeno, pero su importancia a este respecto no es clara aún. Los andrógenos suprarrenales son metabolizados a compuestos 17-ceto, que se excretan en la orina como 17-cetoesteroides (17-KS).
Aldosterona El principal mineralocorticoide producido por la corteza suprarrenal es la aldosterona, un 18 oxicorticoide producido en la capa glomerular a partir de sus precursores, acetato y colesterol.3 La angiotensina constituye el estímulo dominante para la biosíntesis de aldosterona [ver figura 3]; tanto la angiotensina como la ACTH aumentan la secreción de aldosterona al acelerar la conversión de colesterol a D5-pregnenolona. Otros estímulos para la secreción de aldosterona incluyen el aumento del potasio sérico, la pérdida de volumen sanguíneo, la disminución del sodio sérico y el aumento en los niveles de estrógenos. Existen evidencias de que la liberación de aldosterona es inhibida por mecanismos dopaminérgicos. La aldosterona circula libremente y es degradada enzimáticamente durante la depuración hepática, así como por conjugación y excreción renal. Su grado de depuración metabólica depende estrechamente del aporte sanguíneo hepático. La disminución de éste, como ocurre en diversos trastornos hepáticos, puede producir hiperaldosteronismo transitorio; las interacciones con otros mecanismos que controlan la secreción de aldosterona determinan si esta alteración perdura.
REGULACION DE LA SECRECION SUPRARRENAL
La ACTH es el principal regulador de la producción de cortisol y andrógenos suprarrenales [ver figura 3].3 A su vez, la síntesis y liberación de ACTH por la hipófisis es controlada sobre todo por la hormona liberadora de corticotropina (CRH, por sus siglas en inglés, n. el t.), un péptido de 41 aminoácidos producido en el hipotálamo.4
La vasopresina, también llamada arginina vasopresina (AVP) u hormona antidiurética (HAD), también participa en la regulación basal de la ACTH.5,6 Aunque la vasopresina, la oxitocina y las catecolaminas influyen en el ciclo diurno de ACTH y cortisol, el principal determinante de este ciclo parece ser la liberación diurna de CRH. Durante los estados tensionales aumenta de dos a 10 veces la producción de esteroides suprarrenales; esta respuesta está en relación con un efecto potencial, tanto de la CRH como de la HAD, más que un simple efecto aditivo.
La ACTH se sintetiza como un pequeño segmento de la compleja molécula llamada pro-opiomelanocortina (POMC).l0 El producto primario del genes una molécula compleja que también incluye las secuencias de aminoácidos para la b-lipoproteína (b-LPH), un péptido de unión, las hormonas estimuladoras de los melanocitos µ y b (µ-MSH y b-MSH), la b-endorfina, la µ-LPH, la metencefalina y al menos dos fragmentos adicionales, el péptido del lóbulo intermedio parecido a la corticotropina (CLIP, por sus siglas en inglés, n. del t.) y una secuencia de 16 kilobases (16 kb) de significado fisiológico no determinado [ver figura 4]. La POMC también se produce en tejidos no hipofisarios, pero su procesamiento postraducción difiere en varios aspectos. En la hipófisis una hendidura separa al fragmento más largo al principio de la ACTH y ambas porciones son glucosiladas; más tarde, los fragmentos más pequeños son liberados. Aunque la liberación de la b-lipoproteína tiende a ser paralela a la de ACTH, la función de esta molécula y la de los otros dos fragmentos del precursor mayor no está completamente aclarada.
Al igual que otras hormonas hipofisiarias, la ACTH se secreta en pulsos.7 Ocurren de uno a tres pulsos cada hora, con una concentración mayor por la mañana. Debido a que el cortisol resultante desaparece más lentamente que la ACTH, los niveles matutinos iniciales se suman al ritmo diurno, que es mayor durante la mañana y menor por la tarde. Además de la producción de pulsos endógenos, la liberación de ACTH y cortisol también es estimulada por la fiebre, la hipoglucemia, las tensiones de todo tipo, los trastornos psicológicos y la disminución en los niveles de cortisol plasmático. El factor dominante en la inhibición de ACTH es el nivel plasmático de cortisol. Todos los glucocorticoides sintéticos tienen el mismo efecto, que es mediado por la supresión de la transcripción de ARN mensajero (ARNm) para la síntesis de POMC.8 Se piensa que una supresión similar por los glucocorticoides inhibe la producción de CRH. Por lo tanto, el cortisol tiene un efecto de retroalimentación negativa a nivel tanto hipota lámico como hipofisiario [ver figura 3].
Pruebas de función suprarrenal
Las principales pruebas utilizadas para evaluar la función suprarrenal son la determinación en plasma de ACTH y cortisol y de 17-OHS urinarios y cortisol libre en orina de 24 horas.
Determinación de cortisol y ACTH en plasma Los valores basales de cortisol plasmático fluctúan entre 10 y 25 µg/100 ml por la mañana y 2 y 10 µg/ml durante la noche. El nivel de cortisol en plasma varía por los cambios en la cantidad de globulina fijadora de cortisol, que suelen ser paralelos a los de la globulina fijadora de tiroxina. Por ejemplo, ambas proteínas se elevan por el estímulo estrogénico y disminuyen en las enfermedades hepáticas. Los niveles basales de cortisol suelen ser menos importantes que su respuesta a los estímulos fisiológicos; por lo tanto, los niveles de cortisol plasmático habitualmente se miden después de la administración de un esteroide exógeno para determinar si el eje hipófisis-suprarrenal es susceptible a la inhibición por retroalimentación, es decir, si puede suprimirse, y después de la administración de ACTH para observar si la suprarrenal responde a la estimulación.
A continuación se describen algunas pruebas bien estandarizadas para evaluar la función suprarrenal:
1. Para determinar la supresión del eje hipófisis suprarrenal, se administra 1 mg de dexametasona (un análogo del cortisol) por vía oral a las 11 :P.M., y se cuantifican los niveles de cortisol plasmático a las 8:00 A.M. del día siguiente. Los valores normales son menores de 5 µg/100 ml.
2. Para determinar la respuesta suprarrenal, se administran por vía intravenosa 0.25mg de cosintropina (ACTH sintética). Se mide el cortisol plasmático a los 0 y 60 minutos; un valor de 20 µg/dl en cualquier momento de la prueba indica respuesta suprarrenal normal.
3. Para evaluar la reserva hipófisis-suprarrenal, se administran 3 g de metirapona por vía oral a media noche, y se determinan los niveles plasmáticos de cortisol y desoxicortisol (compuesto S) a las 8:00 A.M. del día siguiente. La metirapona inhibe la 11-hidroxilación, lo que ocasiona disminución en los niveles de cortisol y aumento de ACTH. Si la reserva hipófisis-suprarrenal está intacta, los niveles de cortisol en plasma deben ser menores de 5 µg/100 ml y los de desoxicortisol mayores de 10 µg/100 ml.
4. Un método alternativo para evaluar la reserva hipófisissuprarrenal consiste en la administración intravenosa de insulina, 0.15U/kg. La hipoglucemia secundaria debe provocar respuestas tanto de cortisol (normal, 20 µg/dl) como de hormona del crecimiento (> 8 ng/ml). Esta prueba debe realizarse en presencia de un médico preparado para administrar 25 ml de glucosa al 50 porciento por vía intravenosa en caso de una reacción de hipoglucemia.
5. Para determinar la reserva hipofisiaria de ACTH se puede administrar CRH ovina en dosis de 1 µg/kg de peso corporal. Tanto los niveles de ACTH como los de cortisol en plasma se elevan en los individuos normales. Esta prueba puede realizarse a cualquier hora del día, pero su sensibilidad es mayor por la tarde.
La ACTH en plasma puede determinarse por prueba de inmunorradiometría (IRMA), que emplea anticuerpos contra las porciones N-terminal y C-terminal de la hormona. A diferencia de la prueba de radioinmunoensayo, la IRMA puede distinguir en forma confiable los niveles bajos, normales y elevados de ACTH.9
Niveles de 17-hidroxiesteroides urinarios Aunque tiene la ventaja de integrar la secreción de cortisol durante un periodo de 24 horas, la utilidad de la determinación de 17-OHS urinarios es limitada, ya que éstos se elevan en la obesidad y disminuyen en las enfermedades hepáticas y renales. Por otra parte, la recolección adecuada de la muestra urinaria es difícil para muchos pacientes.
Cortisol libre en orina de 24 horas Esta prueba también requiere que la recolección de la muestra se realice de forma adecuada. Los valores en individuos obesos rara vez se superponen con los de aquellos pacientes con enfermedad de Cushing, y los trastornos hepáticos no alteran los resultados. La excreción urinaria de cortisol libre se ha convertido en un valioso indicador de hiperfunción suprarrenal. Es menos seguro para detectar insuficiencia suprarrenal.l0
Hiperfunción de la corteza suprarrenal: Síndrome de Cushing
El síndrome de Cushing es un estado poco frecuente de hiperfunción de la corteza suprarrenal. Sin embargo, es importante clínicamente por dos razones. En primer lugar, porque es a menudo curable. En segundo lugar, porque el exceso de glucocorticoides es una causa potencial de los problemas que el médico observa cotidianamente, como obesidad, hipertensión arterial, diabetes mellitus, litiasls renal, osteoporosis, púrpura, trastornos mentales, alteraciones menstruales, impotencia, hirsutismo, edema, hipocalemia, susceptibilidad a las infecciones y cicatrización deficiente. Aunque ninguno de estos poblemas hace pensar per se en el diagnóstico de hipercortisolismo, la coexistencia de varias características clínicas pueden justificar la investigación de síndrome de Cushing. Entre las más útiles se encuentran la facies pletórica, las equimosis, la presencia de hipocalemia de causa desconocida, los defectos en la cicatrización y los cambios en las facciones, basándose en la comparación del aspecto actual con una fotografía previa.1l
Las causas del síndrome de Cushing son el exceso de ACTH, el aumento en la producción de cortisol por las suprarrenales y la ingestión iatrogénica o facticia de corticoides.l2
EXCESO DE ACTH
Producción hipofisaria excesiva (Enfermedad de Cushing)
El exceso de ACTH hipofisaria con hiperplasia suprarrenal bilateral es la causa de casi dos tercios de los casos de síndrome de Cushing. Esta enfermedad es más frecuente en mujeres en edad reproductiva y su aparición suele ser paulatina. Cualquier síntoma producido por el exceso de cortisol puede aparecer inicialmente, pero finalmente ocurren trastornos menstruales en las mujeres, impotencia en los hombres, aparición de equimosis, facies de luna llena y plétora generalizada. La secreción de andrógenos suprarrenales está aumentada, por lo que en las mujeres son frecuentes el hirsutismo y la alopecia leve. El exceso de andrógenos puede ejercer cierto efecto protector contra la influencia antianabólica del cortisol, pero la gravedad de las manifestaciones clínicas es más o menos proporcional a la duración del trastorno y a la cantidad de ACTH secretada. La pigmentación, una manifestación del exceso de ACTH, puede ser un dato de gran valor para el diagnóstico, aunque este signo puede ser difícil de reconocer, sobre todo en individuos de piel oscura.
En teoría, puede producirse un exceso de ACTH hipofisaria por un aumento de la hormona liberadora de corticotropina o por una neoplasia primaria de la hipófisis. Cushing consideraba que la mayoría de los casos eran causados por un tumor hipofisario y en la actualidad la enorme experiencia de curación secundaria a la extirpación de microadenomas de hipófisis apoya esa interpretación. Los estudios moleculares han indicado que la mayoría de los microadenomas productores de ACTH son monoclonales.13 Como en el caso de otros tumores, esto es una fuerte evidencia de que el origen de la neoplasia es hipofisiario, más que una respuesta hiperplásica a CRH. Sin embargo, la recurrencia de los tumores después de una aparente mejoría y la respuesta de algunos pacientes a fármacos como la bromocriptina, agonista de la dopamina, apoyan, aunque no demuestran, un origen hipotalámico. Por el contrario, el éxito de la radioterapia hipofisaria, especialmente en niños, apoya la opinión de que, como afirmó Cushing, la enfermedad de Cushing es con más frecuencia un hiperadrenalismo secundario a tumor hipofisario.
Los siguientes datos son característicos de la enfermedad de Cushing:
1. Elevación de los niveles de cortisol libre urinario, por lo general mayores de 150 µg/24 horas.
2. Pérdida del ritmo circadiano de la ACTH y el cortisol plasmáticos: los niveles vespertinos de cortisol son mayores de 10 µg/dl; la excreción urinaria de 17-OHS también pierde su ritmo diario.
3. Pérdida de la capacidad para suprimir la secreción de ACTH con dosis fisiológicas de glucocorticoides: la administración de 1 mg de dexametasona a media noche no impide la elevación matutina de los niveles de cortisol por encima de 10 µg/dl.
4. Inhibición de la secreción de cortisol cuando se administran dosis mayores de dexametasona, 2 mg cada seis horas durante dos días: los niveles urinarios de 17-OHS y 17-KS disminuyen a menos del 50 porciento de las cifras control.
Producción ectópica de ACTH
Existen dos variantes de síndrome de Cushing causado por la producción ectópica de ACTH (no hipofisiaria). La primera es el resultado de la producción intensa de ACTH por neoplasias malignas, siendo las más frecuentes las de pulmón, timo, páncreas y riñón.14 El síndrome consiste en hipercorticismo agudo, en el que predominan los efectos mineralocorticoides secundarios a la secreción de grandes cantidades de cortisol, más que los cambios inducidos por su acción glucocorticoide. La alcalosis hipocalémica, la debilidad y la pérdida de peso son los elementos clínicos sobresalientes y el aspecto cushinoide es poco frecuente. El cortisol plasmático y los esteroides urinarios se encuentran a menudo mucho más elevados y el paciente rara vez tiene una franca apariencia cushinoide. Además, los niveles urinarios o plasmáticos de 17-OHS no se suprimen por una dosis diaria de 8 mg de dexametasona. Por tanto, el dato principal para establecer el diagnóstico de este tipo de producción ectópica de ACTH es la discrepancia entre un cuadro clínico poco llamativo y la presencia de niveles muy elevados en la secreción de esteroides. El gran efecto mineralocorticoide en este padecimiento es consecuencia de la gran producción de cortisol y de la saturación de las dos vías de inactivación del mismo (la conversión acortisona por la 11-hidroxiesteroide deshidrogenasa y la reducción del anillo A a dehidrocortisol). Como consecuencia se acumula cortisol, que se une al receptor de los mineralocorticoides y causa retención de sodio con caliuresis.15,16
La segunda forma de síndrome de Cushing ectópico se relaciona con lesiones benignas o malignas como carcinoides bronquiales o tímicos. El cuadro clínico en estos casos es indistinguible del síndrome de Cushing secundario a alteraciones hipofisarias y son necesarios estudios especiales para hacer la diferenciación [ver adelante, Diagnóstico].
Producción ectópica de CRH
La producción ectópica de CRH por tumores puede estimular una producción suficiente de ACTH en la hipófisis para ocasionar hiperplasia suprarrenal y síndrome de Cushing. El tumor más implicado en esta producción ectópica de CRH es el carcinoide bronquial, y la mayor parte de estos tumores secretan también ACTH.1 El cuadro clínico puede ser típico de enfermedad de Cushing o presentar las manifestaciones del síndrome de secreción ectópica de ACTH como hipocalemia, alcalosis y debilidad.17 La prueba con dexametasona en dosis altas y bajas no muestra inhibición de la función hipofisiaria-suprarrenal, y los niveles de ACTH en plasma se encuentran muy elevados.
EXCESO DE CORTISOL SUPRARRENAL
Las neoplasias suprarrenales hipersecretoras de cortisol constituyen menos de la tercera parte de los casos de síndrome de Cushing espontáneo. Aunque los síntomas varían, hay ciertos datos clínicos que son útiles para el diagnóstico. Los adenomas suprarrenales benignos a menudo secretan sólo cortisol, por lo que las manifestaciones clínicas reflejan el exceso de cortisol sin aumento de andrógenos. Debido a que el cortisol suprime la secreción endógena de ACTH, otras funciones suprarrenales se en cuentran disminuidas y los 17-cetosteroides tienden a ser bajos. Los carcinomas suprarrenales que producen cortisol en exceso casi siempre producen grandes cantidades de andrógenos; en la mujer aparece hirsutismo y virilización y los cetosteroides urinarios tienden a ser muy altos (50 mg/ 24 horas).l8 Otras características diagnósticas que indican la presencia de tumores suprarrenales incluyen una amplia variabilidad en la actividad secretora, la falta de supresión por altas dosis de dexametasona, alteraciones en la urografía excretora (UE) en el lado afectado, y una masa palpable en cerca del 50 por ciento de los casos de carcinoma suprarrenal.
La menos frecuente y más enigmática causa de síndrome de Cushing es la hiperplasia nodular bilateral no dependendiente de ACTH. Se han postulado varios mecanismos raros, incluyendo mutaciones activadoras de los receptores de proteína G que estimulan a la adenilato ciclasa.19 La hiperplasia macronodular es muy probable que se presente como una complicación tardía de la enfermedad de Cushing no tratada, en la cual uno o más nódulos se hacen autónomos. Dependiendo de los niveles de producción autónoma del cortisol, la ACTH hipofisaria puede estar elevada o aparentemente normal; las pruebas con dosis altas de dexametasona deben inhibir a la ACTH pero no el cortisol. Por lo menos en un caso se comprobó la progresión de dependencia hipofisaria a la autonomía suprarrenal.20
La causa más confusa de síndrome de Cushing es la displasia suprarrenal micronodular bilateral, trastorno que en ocasiones es de tipo familiar y en el que los niveles de cortisol están elevados y los de ACTH bajos, observándose en las suprarrenales nódulos pequeños activos y áreas de tejido atrófico.21 En algunas familias la hiperplasia suprarrenal parece deberse a una inmunoglobulina estimuladora de las suprarrenales, un autoanticuerpo hacia los receptores de ACTH similar a las inmunoglobulinas estimuladoras del tiroides en la enfermedad de Graves.
La causa menos frecuente de hiperplasia suprarrenal nodular es el hipercortisolismo dependiente de alimentos, en el que la expresión inadecuada del receptor del polipéptido inhibitorio gástrico (PIG) por las células suprarrenales provoca una secreción exagerada de cortisol en respuesta a los alimentos (pero no en la mañana). Este tipo recién conocido de síndrome de Cushing podría ser muy importante y pueden implicarse mecanismos iguales o parecidos en otros trastornos.22-24
Más adelante se discute el exceso de cortisol por ingestión iatrogénica o facticia [ver adelante, Uso farmacológico de los glucocorticoides].
DIAGNOSTICO
Por las diversas manifestaciones del exceso de los glucocorticoides, es evidente la utilidad de contar con una prueba simple y sencilla que excluya el diagnóstico de síndrome de Cushing en un paciente con pocas probabilidades de tenerlo. Para este fin la prueba más adecuada es la cuantificación de cortisol plasmático matutino después de administrar 1 mg de dexametasona la noche anterior. Si el cortisol se encuentra por debajo de 5µg/dl, se descarta el síndrome de Cushing. Si los valores se encuentran por encima de esta cifra y el paciente no está recibiendo tratamiento estrogénico, está indicada la evaluación completa y definitiva para establecer el diagnóstico. Esta prueba no confirma el diagnóstico en un paciente con probabilidad de síndrome de Cushing. Debido a la gran prevalencia de obesidad cushinoide (quizá 3,000 casos por millón en adultos), la ausencia de supresión por la dexametasona en estos pacientes tiene muy poco significado a menos que la evidencia clínica de síndrome de Cushing sea muy fuerte.25
Cuando se sospecha clínicamente el síndrome de Cushing, se puede confirmar mediante la prueba con 1 mg de dexametasona o la determinación del cortisol libre urinario en 24 horas. Debido a que la prueba en plasma se asocia con más falsas positivas, se usa más la determinación de cortisol en orina. Cuando existen dudas acerca del diagnóstico se recomienda realizar ambas pruebas.
Si la prueba de escrutinio fue positiva, la evaluación definitiva debe incluir lo siguiente [ver figura 5]:
l. Administración de dexametasona, 0.5 mg cada seis horas durante dos días. Los niveles de l7-OHS en orina de 24 horas menores de 3.5 mg en el segundo día o cifras de cortisol plasmático inferiores a 5 µg/dl en la mañana del tercer día descartan el síndrome de Cushing; los niveles mayores lo confirman. Debe enfatizarse la importancia de que este punto quede totalmente claro. Aunque parezca irónico, las pruebas para localizar la causa patológica del síndrome de Cushing no son útiles para establecer su presencia. Por lo tanto, debe demostrarse sin duda alguna que existe hipercortisolismo antes de realizar más estudios.
2. El primer paso para establecer la causa consiste en distinguir entre el hipercortisolismo dependiente de ACTH y el independiente. Para ello deben realizarse varias mediciones plasmáticas de ACTH [ver figura 6] con determinación simultánea de cortisol. Los nuevos estudios inmunorradiométricos para ACTH permiten diferenciar con claridad los valores bajos y suprimibles de los tumores adrenales (< 5 pg/ml) de los valores aparentemente normales o elevados de los casos dependientes de ACTH.26 Un nomograma elaborado en la Clínica Mayo puede identificar el origen patológico del hipercortisolismo en alrededor del 90 porciento de los casos midiendo el cortisol en plasma, el cortisol libre en orina y los niveles de ACTH.27
3. Administración de dexametasona, 2 mg cada seis horas durante dos días. Los siguientes resultados son característicos de exceso de ACTH hipofisiaria (enfermedad de Cushing): un nivel de 17-OHS en orina el segundo día menor del 35 porciento del basal, un nivel de cortisol libre urinario el segundo día menor del 10 porciento del basal o una concentración de cortisol en plasma la tercera mañana menor de 10 µg/dl.28 El fracaso para llegar a estos resultados indica una fuente no hipofisiaria de ACTH o un tumor o hiperplasia suprarrenales autónomos. El valor de la ACTH determinada en el inciso 2, mencionado antes, permitirá esta distinción. La prueba con dosis altas de dexametasona se ha modificado para evitar la incomodidad e inexactitud de la recolección urinaria. Se obtiene una determinación de la concentración basal de cortisol a las 8 A.M., se administran 8 mg de dexametasona por vía oral a las 11 P.M. y se obtiene una segunda medición de cortisol a las 8 A.M. del día siguiente. Un valor de menos del 50 por ciento del basal el segundo día indica origen hipo fisiario,10 pero la sensibilidad y especificidad de esta prueba para hipercortisolismo dependiente de ACTH es mayor si se combina con la prueba estándar de supresión con dosis altas (8 mg/día).29
4. Si los niveles de ACTH son normales o elevados, la tomografía computada (TC) o la resonancia magnética (IRM) de la hipófisis pueden indicar la presencia de un tumor. Si la ACTH está inhibida, se deberá realizar una TC o IRM del área suprarrenal.
5 Los pasos anteriores consiguen un diagnóstico definitivo en el 90 porciento de los pacientes. Los resultados endócrinos y de imágenes se combinan para establecer un origen hipofisiario o ectópico de la ACTH o un tumor suprarrenal como la causa del hipercortisolismo. El problema más difícil de solucionar consiste en distinguir entre la producción de ACTH hipofisaria excesiva y la producción ectópica por alguna variedad de carcinoide [ver antes, Producción excesiva de ACTH].30 Si no se detecta un tumor hipofisario se recomienda realizar una TC del tórax, ya que el 90 porciento de los tumores benignos productores de ACTH y CRH se localizan en esa área.
6.Para los pacientes ocasionales en quienes la distinción de la patología sigue siendo un problema, pueden utilizarse dos estudios recientes. La inyección por vía intravenosa de hormona liberadora de corticotropina ovina (CRHo), 100 µg o l µg/kg provoca una elevación del ACTH y del cortisol plasmático en la enfermedad de Cushing, pero no en el hipercorticismo independiente de ACTH. Sin embargo, todavía persiste cierta sobreposición, en particular con algunos pacientes con producción ectópica de ACTH que responden a esta prueba.31-34
7. La prueba más sofisticada y difícil desde el punto de vista técnico para identificar microadenomas demasiado pequeños para ser visualizados es la administración simultánea de CRHo (i.e., estimulación hipofisaria) y la toma de muestras de ambos senos petrosos inferiores.35 La concentración dos veces mayor de ACTH entre las venas petrosas y las venas periféricas sugiere un adenoma hipofisario y justifica la exploración transesfenoidal. La concentración dos veces mayor de ACTH entre ambas venas petrosas y las cifras basales o después de la administración de CRH se considera una justificación para realizar hemihipofisectomía si no se identifica quirúrgicamente un microadenoma.
Un enfoque nuevo y elegante para localizar producción ectópica de ACTH consiste en el uso de ocreótido radiomarcado. Este análogo de larga acción de la somatostatina se fija a los receptores expresados en los tumores neuroendócrinos como los carcinoides bronquiales, pero no a los adenomas hipofisiarios habituales. Por lo tanto, puede usarse para diagnosticar, localizar y tratar la producción ectópica de ACTH.36,37
Cuando se realizan y correlacionan cuidadosamente, las pruebas descritas deben proporcionar la interpretación definitiva en casi todos los casos de síndrome de Cushing [ver figura 6]. Sin embargo, es indispensable empezar por comprobar que el paciente tiene hipercortisolismo. A menos que el cortisol libre en orina esté elevado, es probable que el examen diferencial y de localización sea confusa e inútil.
Es conveniente considerar algunos aspectos útiles respecto a la evaluación del eje hipófisis-suprarrenal. Algunas circunstancias pueden alterar el ritmo circadiano de la secreción de ACTH y cortisol, produciendo resistencia a la supresión hipofisaria con 1 mg de dexametasona. La más evidente es el estado tensional agudo, como el provocado por la hospitalización o una intervención quirúrgica; los niveles vespertinos de cortisol después de los primeros días de estancia hospitalaria pueden llegar a estar por encima de 10 µg/dl sin acompañarse de datos clínicos anormales. Una segunda causa de dificultades es la depresión y agitación que pueden simular alteraciones en las pruebas dinámicas compatibles con síndrome de Cushing por exceso de secreción de ACTH hipofisaria. En pacientes con depresión ansiosa se pierde el ritmo circadiano y la capacidad de supresión con dexametasona. Esta situación constituye un problema especial porque el hipercorticismo a su vez puede causar depresión ansiosa. El diagnóstico diferencial se confirma por el examen clínico para detectar la presencia de signos del síndrome, y cuantificando las cifras de cortisol libre urinario, que es normal en el deprimido. En el paciente deprimido con nivel urinario de cortisol elevado puede ser útil la prueba de CRH.38
Las dos enfermedades más difíciles de diferenciar del síndrome de Cushing son la obesidad y el alcoholismo; en estas tres afecciones se puede encontrar plenitud facial, debilidad, fatiga y equimosis. En ocasiones los resultados de las pruebas de escrutinio en individuos obesos son anormales, pero las pruebas dinámicas finalmente permiten distinguir a estos sujetos de los que tienen síndrome de Cushing. En el seudosíndrome de Cushing que ocurre en los alcohólicos, las pruebas iniciales pueden rnostrar resistencia a la supresión del cortisol plasmático con la administración de 1 mg de dexametasona y pérdida del ritmo circadiano. Sin embargo, con la abstinencia los valores de cortisol plasmático vuelven a la normalidad y reaparece la capacidad de supresión con dexametasona.39
El hipercortisolismo cíclico o periódico es otra causa de hallazgos de laboratorio confusos e inquietantes en el síndrome de Cushing. Los ciclos pueden variar entre días a meses y pueden persistir durante años. Se requiere de gran cuidado antes de recomendar el tratamiento quirúrgico.40
TRATAMIENTO
El tratamiento de elección en adultos con síndrome de Cushing es la cirugía, y en la forma más común del mismo, la enfermedad de Cushing (i.e., exceso de la producción hipofisaria de ACTH), la micro adenectomía transesfenoidal es el tratamiento de elección.41-45 Si las imágenes de la hipófisis (la IRM es la técnica de mayor sensibilidad) o la prueba del seno petroso identifican un microadenoma [ver figura 7], las cifras de curación son tan altas como del 90 porciento. Ocurren recurrencias en el 5 al 20 porciento de los casos.46 Se obtienen mejores resultados cuando la silla turca es radiológicamente normal. Cuando hay alteraciones en la silla turca o el adenoma es mayor de 10 mm de diámetro, las oportunidades de curación son claramente menores. Se han observado complicaciones como diabetes insípida transitoria y meningitis, pero son fáciles de tratar.
Cuando la microadenectomía tiene éxito los pacientes muestran niveles plasmáticos de cortisol normales o bajos pronto después de la cirugía; dichos pacientes suelen presentar supresión de la función hipofisaria y suprarrenal y pueden necesitar tratamiento sustitutivo durante periodos tan largos como seis meses después de la cirugía.47 Cuando los hallazgos quirúrgicos son poco definidos o cuando los niveles de cortisol no bajan, se puede administrar una inyección de CRH para demostrar la persistencia de células productoras de ACTH. Otras técnicas incluyen tratamiento con partículas pesadas, implante de itrio y radiaciones externas. Todos estos tratamientos son inferiores a la adenomectomía, aunque es importante señalar que, en la mayoría de las instituciones, la radiación externa es el tratamiento de elección en niños.
El entusiasmo actual por el enfoque directo a la hipófisis no debe menospreciar la utilidad ocasional de la adrenalectomía bilateral. En aquellos pacientes con enfermedad grave, complicaciones que ponen en peligro la vida, tumores hipofisarios muy grandes o hiperplasia nodular y en los que el tratamiento hipofisario ha fracasado, la suprarrenalectomía puede ser preferible a una microadenectomía transesfenoidal.48 El tratamiento preoperatorio con metirapone puede modular el hipercortisolismo, incluyendo las alteraciones psicológicas, lo que hace que la cirugía sea más segura. Todos los pacientes manifiestan insuficiencia suprarrenal después de la suprarrenalectomía y un porcentaje importante desarrollan síndrome de Nelson. El autotransplante de tejido suprarrenal se ha utilizado con el fin de evitar la insuficiencia suprarrenal, pero pocas veces se ha logrado implantar una cantidad de tejido suprarrenal que evite la hipofunción y que no ocasione recurrencia del trastorno.
Existen varios tratamientos farmacológicos para el síndrome de Cushing. La aminoglutetimida inhibe la síntesis de cortisol al bloquear la conversión del coleterol a D5- pregnenolona. El mitotane es un agente adrenolítico que inhibe la síntesis de cortisol, y el metirapone reduce la producción de cortisol al inhibir la 11-hidroxilasa. Sin embargo, la toxicidad por la aminogutetimida y el mitotane es alta; incluso cuando ambos medicamentos se administran en combinación para disminuir su toxicidad individual, los resultados rara vez son satisfactorios. Por lo tanto, la utilidad de los fármacos en el síndrome de Cushing se limita a situaciones especiales, como el control de manifestaciones severas antes de lacirugía, como complemento de la remisión parcial inducida por irradiación hipofisaria, o para aliviar los síntomas en enfermos con carcinoma metastásico inoperable o con producción ectópica de ACTH.
La resección quirúrgica es el tratamiento de elección para el carcinoma o el adenoma suprarrenal. Si la extirpación del tumor tiene éxito, se presenta insuficiencia suprarrenal y el paciente necesita tratamiento con corticoesteroides en forma temporal. La recuperación de la función normal hipotalámica, hipofisaria y suprarrenal puede retrasarse por periodos tan prolongados como dos años.49
Síndrome de Nelson
Del 10 al 40 porciento de los pacientes con enfermedad de Cushing (i.e., exceso de ACTH hipofisaria) que son tratados con suprarrenalectomía bilateral desarrollan síndrome de Nelson.50 Este síndrome, caracterizado por hiperpigmentación notable y un tumor cromófobo de la hipófisis, puede desarrollarse entre seis meses y I5 años después de la suprarrenalectomía. En algunos casos el crecimiento local agresivo de la tumoración causa oftalmoplejía, defectos visuales y, en ocasiones, infarto hipofisario. Debido a que se desconoce qué características del síndrome de Cushing anteceden al desarrollo del síndrome de Nelson, los pacientes tratados con suprarrenalectomía bilateral deben vigilarse a intervalos regulares. El diagnóstico se confirma mediante la demostración de hiperpigmentación, agrandamiento de la silla turca y niveles elevados de ACTH.
Las concentraciones de ACTH en plasma después de la adrenalectomía para el síndrome de Cushing tienden a ser mayores a las observadas en la enfermedad espontánea de Addison y con frecuencia aumentan 20 veces lo normal, hasta 300 a 1,000 pg/ml. Es probable, aunque no seguro, que la determinación anual de los niveles de ACTH permitan detectar la enfermedad antes de que se manifieste clínicamente. El tratamiento incluye resección quirúrgica, radioterapia con partículas pesadas, ciproheptadina y bromocriptina. Los informes difieren con respecto a si la irradiación profiláctica de la hipófisis evita el desarrollo del síndrome de Nelson posadrenalectomía.
Uso farmacológico de los glucocorticoides
Parael clínico, los glucocorticoides han sido fármacos de gran valor que a la vez le han ocasionado serios problemas. Aunque su uso en ciertas enfermedades llega a salvar la vida, producen síndrome de Cushing en algunos pacientes y supresión del eje hipotálamo-hipófisis-suprarrenal en todos los enfermos sometidos a tratamiento crónico.5l Ciertas complicaciones son características del sindrome de Cushing iatrogénico, incluyendo hipertensión intracraneana benigna, glaucoma, cataratas y pancreatitis.5l
Aunque rara vez se ha demostrado que la supresión del eje hipotálamo-hipófisis-suprarrenal cause la muerte, sigue siendo preocupante en los sujetos tratados con glucocorticoides. Es imposible determinar el tiempo mínimo de tratamiento necesario para que se establezca la supresión de la función endógena; si el tiempo lo permite, puede realizarse la prueba corta con ACTH. Se administra una dosis de 0.25 mg de cosintropin por vía intravenosa y, si el cortisol en plasma a los 30 minutos es mayor de 18 ng/dl, el eje hipotálamo-hipófisis se considera normal. Sin embargo, con frecuencia no hay tiempo para esperar el resultado, por lo que es conveniente administrar suplementos de esteroides a cualquier paciente tratado antes con estos fármacos. La revisión de la historia y la experiencia con la insuficiencia suprarrenal secundaria han sugerido que los suplementos farmacológicos con esteroides son excesivos y que la dosis debe depender de dos aspectos: la dosis recibida por tiempo prolongado y la magnitud de la enfermedad o cirugía. En casos de cirugía menor es adecuado administrar 25 a 50 mg/día de hidrocortisona o su equivalente, para situaciones intermedias 50 a 75 mg/día y para cirugía mayor 100 a 150 mg/día durante uno a tres días.52 Esta dosis puede disminuirse en un 50 porciento cada día, comenzando dos días después de la cirugía si todo va bien.
La recuperación total de la supresión intensa del eje hipotálamo-hipófisis-suprarrenal puede tardar hasta dos años después de la suspensión del tratamiento, y no parece existir manera de acelerar este proceso. Durante este periodo los pacientes pueden manifestar síntomas de supresión a pesar de tener niveles basales normales de cortisol, así como de cierta respuesta de la hipófisis ante el estímulo. Para valorar la recuperación, la mejor prueba parece ser la respuesta del cortisol plasmático ante el estímulo con ACTH o a la hipoglucemia inducida por insulina.53 El nivel basal o estimulado mayor de 18 µg/dl indica una respuesta suprarrenal normal. Sin embargo, ninguna prueba es con fiable al 100 porciento. La prueba de estimulación con ACTH sintética puede en ocasiones ser normal en pacientes que responden en forma subnormal al estrés.54
Aldosteronismo primario
El aldosteronismo primario es un síndrome manifestado por hipertensión arterial e hipocalemia que ocurre en el 0.05 a dos porciento de los pacientes hipertensos55. Entre las causas predominan los adenomas unilaterales (en alrededor del 70 porciento de los casos), la hiperplasia bilateral idiopática (en el 20 a 30 porciento) y, en raras ocasiones, un carcinoma adrenocortical.56-58 Los pacientes con aldosteronismo primario se encuentran entre la población hipertensa que tiene una concentración limítrofe de potasio, en el rango de 3.4 a 3.6 mEq/L, o de hipocalemia franca que ocurre en forma espontánea o por el uso de diuréticos leves. En los pacientes que reciben diuréticos éstos deben ser suspendidos durante cuatro semanas y se repletará la deficiencia de potasio. A continuación se administra al paciente una dieta que proporcione por lo menos 100 mEq de sal al día y se miden la aldosterona y la renina en plasma a las 8 A.M. después de que el paciente ha estado recostado toda la noche y de nuevo cuando el paciente ha estado de pie durante cuatro horas. También se mide la concentración de aldosterona en orina de 24 horas. Además de la hipocalemia causada por la carga de sodio, en los pacientes con aldosteronismo primario la concentración plasmática de renina es menor de 1 ng/ml/h, la de aldosterona es mayor de 15 ng/dl y los metabolitos urinarios de la aldosterona estan elevados. Después de localiza el tumor o se confirma la hiperplasia por medio de tomografía computada y por medición de aldosterona y cortisol en ambas venas suprarrenales.59
La hipertensión y la hipocalemia del síndrome de Conn (adenoma unilateral) suelen responder a la suprarrenalectomíaunilateral. Los pacientes con hiperplasia se tratan médicamente. Se ha sugerido que la obtención de una sola relación de actividad plasmática de aldosterona/renina puede usarse como método de escrutinio para el hiperaldosteronismo primario.60,61
La hipertensión e hipocalemia inducidas por la ingestión de orozuz es secundaria al acúmulo de ácido glicirrízico en las células tubulares renales, con la consecuente inhibición de una enzima (la 11 13-hidroxiesteroide deshidrogenasa) que inactiva los mineralocorticoides.16 El aldosteronismo remediable por glucocorticoides (ARG) típicamente se manifiesta en la infancia, y existe gran frecuencia de hipertensión severa con eventos cerebrovasculares entre los adultos jóvenes de las familias afectadas. Como en el síndrome de Conn, la hipocalemia es común, pero no es suficientemente frecuente como para ser útil para el escrutinio. El ARG es un trastorno autosómico dominante causa do por la formación mutante de un gen quimérico en la zona fasciculada cuyo producto proteico combina las activida des de la 11b-hidroxilasa y la sintetasa de aldosterona. Este defecto causa exceso de aldosterona que depende de ACTH en lugar de angiotensina. Al suprimir la ACTH los glucocorticoides disminuyen tanto la hipertensión como la hipocalemia, Como en otros trastornos autosómico dominantes, el escrutinio ha descubierto mucho más individuos afectados de lo que antes de sospechaba.62
Hipofunción de la corteza suprarrenal
La insuficiencia suprarrenal aguda es un trastorno poco frecuente que suele desarrollarse durante una enfermedad grave. La causa más cornún es la hemorragia suprarrenal por anticoagulantes, y parece existir una mayor asociación con la trombocitopenia inducida por heparina y con el síndrome de anticuerpos antifosfolípido.63,64 La insuficiencia también puede presentarse en pacientes con sepsis aguda o después de cirugía, infarto del miocardio o embarazo.65 Los datos clínicos incluyen náusea, debilidad, hipotensión arterial, colapso y dolor abdominal o en la espalda. Este dolor puede ser el único dato que indica la complicación.66-68
El diagnóstico es sencillo, las pruebas sistemáticas de laboratorio probablemente muestren niveles bajos de sodio sérico y niveles elevados de potasio y BUN. Sin embargo, la evaluación definitiva requiere cuantificar los niveles de cortisol plasmático antes y 60 minutos después de la administración de 0.25 mg de ACTH sintética (Cortrosyn). Si al diagnóstico se retrasa excesivamente, se deben administrar 100 mg de hidrocortisona por vía intravenosa al paciente seguidos de 10 mg de cortisol por hora hasta que se obtengan los resultados de las pruebas. En situaciones graves la prueba de ACTH debe diferirse y se debe empezar el tratamiento de inmediato, después de haber tomado la muestra para la determinación del cortisol plasmático. La concentración de cortisol menor de 10 µg/dl cuando la muestra se examina en retrospectiva sugiere por lo menos insuficiencia suprarrenal parcial, un valor por arriba de 18 µLg/dl indica una respuesta normal al estrés. La TC y la IRM pueden confirmar el diagnóstico de hemorragia suprarrenal.
ENFERMEDAD DE ADDISON
La insuficiencia suprarrenal crónica, o enfermedad de Addison tiene dos causas principales. En la actualidad las infecciones granulomatosas crónicas, como la tuberculosis o la histoplasmosis, son la causa de una minoría de los casos en los Estados Unidos. La causa más frecuente es un trastorno autoinmune en el que la invasión suprarrenal por linfocitos y células plasmáticas se acompaña de anticuer pos antisuprarrenales en el plasma. Esta entidad puede aparecer sola o con hipotiroidismo secundario a tiroiditis de Hashimoto [ver adelante, Síndrome de deficiencia poliendócrina]. La insuficiencia suprarrenal puede ocurrir también como manifestación de trastornos hereditarios caracterizados por la degeneración progresiva de la mielina, ya sea en el cerebro (adrenoleucodistrofia) o en la médula espinal (adrenomielodistrofia).
La insuficiencia suprarrenal puede ser una complicación del síndrome de inmunodeficiencia adquirida (SIDA).69 Las suprarrenales se infiltran con patógenos oportunistas y por sarcoma de Kaposi. Aunque se ha observado disminución de la reserva suprarrenal en varias series de pacientes, la insuficiencia suprarrenal típica, confirmada por pruebas y por la respuesta al tratamiento, se ha visto con poca frecuencia en pacientes con SIDA. El tratamiento de pacientes con SIDA con megestrol, un análogo de progestinas con posible efecto anabólico, suprime en ocasiones el eje hipófisis-suprarrenal y causa hipoadrenalismo.70-72
La tomografía computada ha permitido aclarar la etiología de la insuficiencia suprarrenal. Las suprrarrenales aumentadas de tamaño sugieren adrenalitis tuberculosa activa73 o tumor metastásico, cada uno con importantes implicaciones clínicas. Los pacientes en los que se sospeche tuberculosis activa deben recibir tratamiento definitivo. Ha surgido una consecuencia interesante de esta decisión: el tratamiento con rifampicina puede precipitar insu ficiencia suprarrenal en los pacientes con Addison tratados o no tratados.74 Este estado de insuficiencia ocurre porque el medicamento induce enzimas hepáticas que metabolizan esteroides y que causan la depuración acelarada de la secreción endógena subnormal o de las cantidades terapéuticas habituales de hormonas suprarrenales. El anestésico etomidato y el antibiótico antimicótico ketoconazol pueden también inducir insuficiencia suprarrenal reversible, pero su efecto es secundario a un deterioro en la biosíntesis de esteroides.77,76 Los pacientes con metástasis en las suprarrenales pueden presentar insuficiencia súbita, por lo que cuando se encuentra crecimiento de estas glándulas en pacientes con enfermedad metastásica conocida debe considerarse la administración profiláctica de remplazo esteroideo.77
Los pacientes con insuficiencia suprarrenal crónica presentan debilidad, anorexia, pérdida de peso, hipotensión e hipovolemia. Un dato útil para el diagnóstico es la presencia de hiperpigmentación sobre los pliegues de las palmas de las manos y otras áreas corporales, en puntos de presión y alrededor de las aréolas y pezones. Excepto por la tendencia a la hipercalemia, los resultados de los exámenes de laboratorio de rutina no son característicos hasta que ocurre el colapso suprarrenal, caracterizado por hiponatremia, hipercalemia, hipoglucemia, hemoconcentración y elevación del nitrógeno ureico. Las alteraciones en el nitrógeno ureico y los electrolitos son secundarios en gran parte a la deficiencia de volumen y a la azoemia prerrenal. La única prueba diagnóstica especi fica es la determinación de los niveles de cortisol plasmático o de los 17-OHS, antes y después de la estimulación con ACTH . La selección de la prueba diagnóstica dependerá de la valoración del estado clínico del paciente.
Si no parece existir insuficiencia suprarrenal y es necesaria alguna prueba para descartar esta posibilidad, la inyección intravenosa de ACTH sintética, en dosis de 0.25 mg, es sensible y específica. Se determinael nivel plasmático de cortisol antes de la inyección y 60 minutos después. La respuesta normal consiste en un aumento de al menos 18 µg/dl o mayor [ver figura 8]. Sin embargo, si el valor basal es mayor de 20 µg/dl, el aumento puede ser mínimo y la función suprarrenal debe considerarse normal.
En aquellos individuos cuyo cuadro clínico es compatible con insuficiencia suprarrenal (i.e., que tienen hiponatremia o hipotensión), el estudio es un poco diferente. Se realiza una medición matutina de cortisol y ACTH y después, para evitar el colapso suprarrenal, el paciente puede recibir 0.5 mg de dexametasona, y fludrocortisona, 0.1 mg al día, durante la prueba. Se mide el cortisol a los 30 y 60 minutos después de la administración de 0.25 mg de ACTH sintética. La concentración de cortisol en plasma por arriba de 18 µg/dl establece una función suprarrenal normal; sin embargo, si la medición inicial muestra que el cortisol es bajo y sigue bajo con una ACTH por arriba de 100 pg/ml, se establece el diagnóstico de insuficiencia suprarrenal primaria.78 Los enfermos con insuficiencia suprarrenal secundaria a hipopituitarismo pueden diferenciarse de otros sujetos con insuficiencia suprarrenal primaria, o enfermedad de Addison por la presencia de deficiencia de otras hormonas hipofisarias y por un nivel de ACTH , en el límite inferior normal o por debajo del mismo. La insuficiencia suprarrenal secundaria puede confirmarse también administrando metirapone (30 mg/kg por vía oral administrado con los alimentos a media noche). Un nivel en plasma de ll-desoxicortisol por arriba de 7 µg/dl confirma la función normal del eje hipotálamo-hipófisis suprarrenal (HHS). Esa prueba debe hacerse con el pacien te internado por la posibilidad de inducir insuficiencia suprarrenal. En ocasiones la prueba de metirapone descubre una respuesta subnormal en un paciente con prueba normal de ACTH aguda.79 En estos pacientes, en especial en los que tienen menos de seis semanas desde el inicio de la deficiencia suprarrenal, la prueba con ACTH intravenosa puede ser normal porque no ha ocurrido aún atrofia suprarrenal.
En los pacientes con sospecha de crisis suprarrenal no debe permitirse ningún retraso para realizar procedimientos diagnósticos; se toma una muestra de sangre para la determinación del cortisol plasmático y a través de la misma aguja se administra la primera dosis de tratamiento. Es mejor administrar el tratamiento sustitutivo para la insuficiencia suprarrenal aguda con hemisuccinato de hidrocortisona por vía intravenosa, un bolo inicial de 100 mg seguido de 10 mg/hr durante los dos primeros días de la crisis. Para restaurar el volumen circulante es necesario utilizar soluciones mixtas (glucosadas y salinas), expansores del plasma, y en algunos casos sangre total. Debido a que el paciente con enfermedad de Addison suele tener deficiencias considerables de volumen antes de desarrollar la crisis, puede ser necesaria la administración de grandes volúmenes de líquido. Si el estado del individuo mejora puede reducirse la dosis de esteroides de forma gradual, hasta los niveles de mantenimiento, durante tres a cinco días.
El tratamiento sustitutivo para la insuficiencia suprarrenal crónica es de 20 a 25 mg de cortisona al levantarse por la mañana y de 10 a 15 mg por la tarde. Los pacientes también deben recibir fludrocortisona, 0.05 a 0.10 mg por vía oral cada día; los efectos de la dosis deben vigilarse, incluyendo el aumento de peso o la aparición de hipocalemia. La dosis de esteroides deberá incrementarse a discreción, siempre que cualquier individuo con disfunción suprarrenal crónica se exponga a un estado tensional importante. Para una infección de vías respiratorias superiores o influenza es suficiente con duplicar la dosis diaria de cortisona. Sin embargo, si un paciente con enfermedad de Addison sufre vómito, es aconsejable hospitalizarlo para administrar líquidos por vía intravenosa e hidrocortisona.
INSUFICIENCIA SUPRARRENAL SECUNDARIA
Los pacientes con hipopituitarismo que incluye deficiencia de ACTH sufren un síndrome semejante a la enfermedad de Addison excepto porque tienen menor grado de deficiencia de volumen. Debido a que el principal mecanismo de control de la aldosterona es la angiotensina, la regulación del volumen y electrolitos está menos afectada en individuos con panhipopituitarismo que en pacientes con enfermedad de Addison. Los enfermos con deficiencia de cortisol de cualquier causa, incluyendo el pahipopituitarismo, son propensos a la sobrecarga de líquidos y es probable que desarrollen hiponatremia. De manera característica, los niveles de ACTH en plasma están elevados en sujetos con insuficiencia suprarrenal primaria y son normales o no detectables en aquellos con deficiencia de ACTH primaria.
La CRH, actualmente disponible, puede ayudar a diferenciar entre las causas hipotalámicas e hipofisiarias de la insuficiencia suprarrenal secundaria. Si la prueba de intolerancia a la insulina no estimula la producción de cortisol a la hipoglucemia, existe un hipoadrenalismo secundario. Si en dicho paciente la prueba de CRH causa una rápida liberación de ACTH, se sospecha una probable enfermedad hipotalámica; si la CRH no provoca la liberación de ACTH, se supone que la lesión está en la hipófisis.80
SINDROMES POR DEFICIENCIA DE ALDOSTERONA
En ocasiones la deficiencia de aldosterona es la manifes tación única o predominante en la insuficiencia suprarrenal. Los defectos aislados en la biosíntesis de aldosterona son extremadamente raros,81 pero los defectos parciales forman parte del cuadro de hiperplasia suprarrenal congénita secundaria a la deficiencia de 21-hidroxilasa. Los síntomas atribuibles a la deficiencia de aldosterona algunas veces proporcionan los datos iniciales principales para el diagnóstico de insuficiencia suprarrenal; por ejemplo, puede presentarse hipotensión postural, antes que anorexia, pérdida de peso, astenia e hiperpigmentación.
Dos síndromes por deficiencia aislada de aldosterona merecen mención aparte, el hipoaldosteronismo idiopático y el hipoaldosteronismo hiporreninémico.82 El primero es el hipoaldosteronismo idiopático, trastorno muy raro que se presenta como un bloqueo cardiaco secundario a hipercalemia, o como una hipotensión ortostática secundaria a hipovolemia con o sin hiponatremia importante. Sería de esperar que en estos pacientes los niveles plasmáticos y urinarios de aldosterona se encuentren elevados, Junto con un aumento en la actividad de renina plasmática, aunque existen pocos casos publicados con estas características. Para el tratamiento del hipoaldosteronismo idiopático resulta eficaz la fludrocortisona, en dosis de 0.05 a 0.10 mg por día, combinada con la ingestión libre de sal.
El hipoaldosteronismo hiporreninémico es mucho más frecuente que el hipoaldosteronismo idiopático, y en sus formas leves es probable que el diagnóstico pase inadvertido. Los pacientes típicos son mayores de 45 años y padecen insuficiencia renal crónica. El trastorno renal afecta más al tejido tubular e intesticial que a los glomérulos; la diabetes mellitus es una causa frecuente. La característica principal del hipoaldosteronismo hiporreninémico es la hipercalemia crónica, de leve a severa, que en ocasiones se exacerba súbitamente por una hiperglucemia. También suele presentarse acidosis metabólica hiperclorémica, con niveles de sodio normales o bajos. La restricción de la ingesta de sodio exacerba las manifestaciones clínicas. La alteración fisiopatológica de fondo consiste en una concentración baja de renina en plasma a pesar de existir hipercalemia, contracción del volumen circulante e hiponatremia. Aunque la causa del bajo nivel de renina está poco clara, suele atribuirse a un defecto en el aparato yuxtaglomerular. Sin embargo, se ha demostrado que la deficiencia de prostaglandinas inducida por antinflamatorios no esteroides (AINE) puede ser una causa reversible del síndrome; en otros casos, el trastorno se agrava por la administración de heparina, bloqueadores de los canales del calcio o por bloqueadores beta adrenérgicos. El tratamiento del hipoaldosteronismo hiporreninémico incluye la corrección de la acidosis, ingestión libre de sodio y administración cuidadosa de fludrocortisona a las dosis necesarias para controlar la elevación de potasio sérico sin provocar insuficiencia cardiaca congestiva . La heparina es un inhibidor potente de la producción de aldosterona con natriuresis consecuente e hiponatremia ocasional. Ocurre hipercalemia en menos del 10 porciento de los pacientes, pero que puede ser rápida y severa en los enfermos con insuficiencia renal o diabetes mellitus o en enfermos que toman medicamentos que elevan la concentración de potasio sérico.83
Síndromes de deficiencia poliendócrina
Así como la hiperfunción y, las neoplasias pueden afectar a más de una glándula , de la misma forma puede ocurrir con las deficiencias endócrinas. El término síndrome de deficiencia poliendócrina (o síndromes autoinmunes poliglandulares) se refiere a dos tipos de deficiencia endócrina que se superponen, pero que tienen algunas características distintivas.84-86 El tipo I incluye hipoparatiroidismo, hipoadrenalismo y moniliasis mucocutánea, con anemia perniciosa y hepatitis crónica como manifestaciones ocasionales; el inicio en la niñez es característico. El tipo II, también llamado síndrome de Schmidt, incluye insuficiencia suprarrenal, tiroiditis de Hashimoto y, con menor frecuencia, diabetes mellitus insulinodependiente; la enfermedad se inicia a menudo alrededor de los 30 años de edad. Los siguientes hechos plantean una etiología autoinmune: la infiltración linfocítica de las glándulas, el hallazgo frecuente de anticuerpos antiglandulares y la asociación con trastornos autoinmunes conocidos, como la anemia perniciosa y la alopecia. En algunos pacientes ocurre insuficiencia gonadal primaria prematura. Los pacientes con estos síndromes poliglandulares deben vigilarse estrechamente, ya que pueden desarrollar una segunda o tercera alteración endócrina. Así mismo, dado que los síndromes de deficiencia poliendócrina son trastornos familiares, con patrón de herencia de tipo autosómico recesivo,87 es necesario someter a estudio a otros miembros de la familia para descartar la hipofunción endócrina.
Neoplasias suprarrenales e incidentalomas
Una minoría de las neoplasias suprarrenales tienen actividad endócrina.88 Tanto los adenomas como los carcinomas pueden producir síndrome de Cushing y síndromes de virilización. Los carcinomas son típicamente ineficaces en su patrón biosintético. Debido a que los carcinomas suprarrenales pueden sintetizar tipos o cantidades inapropiadas de esteroides, los valores de esteroides urinarios pueden proporcionar la clave para el diagnóstico. Por ejemplo, debe sospecharse cáncer suprarrenal en aquellos pacientes con síndrome de Cushing que presentan una concentración desproporcionadamente alta de 17-KS urinarios en relación con los 17-OHS, o en mujeres con datos de virilización que excretan más de 50 mg de 17-KS al día. Aunque la virilización es poco frecuente en los adenomas benignos, se han publicado varios casos que sólo producían testosterona. Estos casos justifican una mención especial,89-92 porque los niveles urinarios de 17-KS no se encontraban elevados, como suele suceder en las neoplasias suprarrenales que producen hirsutismo, y porque dos de los tumores respondieron a la administración de gonadotropina coriónica aumentando su producción de testosterona. En varios aspectos estos tumores, aunque de origen suprarrenal, se comportan como si se derivaran del tejido gonadal. Las neoplasias feminizantes son las tumoraciones suprarrenales menos frecuentes; deben de considerarse en el diagnóstico de varones con signos de feminización, sobre todo si presentan cifras elevadas de 17-KS, así como de estrógenos93,94
El amplio uso de la TC de abdomen ha dado lugar a un nuevo problema: la detección casual de una masa suprarrenal. Si la lesión es un quiste o un mielolipoma, no se requieren más estudios. Si la lesión es compatible con una masa sólida solitaria, se recomienda un procedimiento de escrutinio económico para detectar un tumor productor de hormonas, Algunos autores investigan solo hiperaldosteronismo y feocromocitoma; sin embargo, el autor considera que el estudio debe incluir la prueba de supresión con 1 mg de dexametasona para el síndrome de Cushing, la determinación de potasio sérico para el aldosteronismo, la medición del sulfato de dehidroepiandrosterona (DHEAS) en plasma o de 17-KS en orina de 24 horas para detectar exceso de andrógenos, y la determinación de ácido vanililmandélico (VMA) en orina de 24 horas para descartar feocromocitoma. La prueba de supresión con dexametasona desenmascara en ocasiones la disfunción en pacientes que no tienen un nivel suprimido de cortisol ni tampoco apariencia cushinoide.95-97 Estos pacientes pueden presentar insuficiencia suprarrenal si se extirpa el adenoma.93 Si se confirma alguna alteración endócrina, está indicada la cirugía. Para las lesiones no funcionantes el tamaño de la lesión es una guía útil.
Las lesiones de menos de 6 cm de diámetro casi siempre son benignas, mientras que las mayores se asocian con tal frecuencia de malignidad que justifica su extirpación.
La aspiración con aguja fina (después de que se ha extirpado el feocromocitoma) puede ser útil. Si se identifica un tumor metastásico puede buscarse el tumor primario. Si se detecta un tumor suprarrenal, aunque la citología no sea concluyente, la combinación de la apariencia más el tamaño de la lesión permitirá una decisión apropiada en relación con la cirugía.99-101
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (HSC) engloba un grupo de trastornos caracterizados por deficiencias enzimáticas hereditarias en la biosíntesis del cortisol; estos defectos tienen como consecuencia aumentos en los niveles de corticotropina, que a su vez producen exceso de andrógenos. En el sexo femenino, el hiperandrogenismo produce masculinización de los genitales externos en la etapa fetal y, si éste no es tratado, ocasiona amenorrea primaria, hipoestrinismo e hirsutismo en la pubertad. En más del 90 porciento de los casos el defecto se encuentra en la enzima 21 -hidroxilasa; con menor frecuencia se afectan la 11-hidroxilasa, la 3-1b-ol-deshidrogenasa, la 17 hidroxilasa y otras enzimas que participan en la síntesis del cortisol. El tratamiento con cortisol produce supresión en la secreción de ACTH, disminuye de los niveles plasmáticos elevados de los precursores de cortisol y corrige las manifestaciones clínicas.
El gen de la deficiencia de la enzima 21-hidroxilasa, junto con un seudogen relacionado, se localiza en el cromosoma 6 dentro del segmento HLA. Varias mutaciones distintas de estos genes han sido identificadas como causa de la deficiencia enzimática que provoca la HSC.102 La incidencia en todo el mundo de deficiencia de 21-hidroxilasa es de alrededor de uno por 14,000 nacimientos, pero existe importante heterogeneidad genética y no todas las manifestaciones son fáciles de explicar.
La deficiencia completa de 21-hidroxilasa suele manifestarse en el recién nacido con virilización e hipertrofia de los genitales. Las expresiones más leves de este padecimiento tienen relevancia clínica en la adolescencia y juventud. Los individuos heterocigotos y homocigotos para el defecto (identificados por estudios genéticos del fenotipo HLA) presentan grados variables de hirsutismo y trastornos menstruales. Por lo tanto, algunos pacientes que presentan hirsutismo, oligomenorrea o incluso síndrome de ovarios poliquísticos idiopáticos manifiestan exceso de andrógenos suprarrenales secundario a defectos parciales en la biosíntesis del cortisol. En los casos en que no hay una virilización congénita el síndrome se denomina hiperplasia suprarrenal congénita de aparición tardía (HSCAT). El método diagnóstico inicial más simple consiste en determinar la 17-OH progesterona (I7-OHP) antes de las 9:00 A.M. durante la fase folicular del ciclo menstrual. Si el nivel de este precursor del colesterol es de 200 ng/dl o menor, el diagnóstico se excluye. Si es de 500 ng/dl o mayor se establece el diagnóstico. Los pacientes con nive les entre estos rangos deben recibir 0.25 mg de ACTH sintética por vía intravenosa y medirse la 17-OHP a los 60 minutos. Los invidividuos afectados tienen concentraciones de 17-OHP mayores de 1,500 ng/dl. Los pacientes con valoresentre l,000 y 1,500 ng/dl pueden ser heterocigotos.103 Existe importante desacuerdo sobre la frecuencia de esta alteración y sobre la prueba adecuada para demostrar las expresiones leves de la enfermedad.104
Una consecuencia práctica de un resultado positivo es que la supresión parcial del eje hipófisis-suprarrenal, como la inducida por la administración de 5 mg de prednisona en el momento de acostarse, puede mejorar la irregularidad menstrual y disminuir el hirsutismo. Las deficiencias par ciales de la 11-hidroxilación y de la 3b-hidroxiesteroide deshidrogenasa también han sido identificadas como posibles variantes de HCSAT que producen hirsutismo. La supresión con prednisona, como se describió antes, es adecuada para tratar a los pacientes con estas deficiencias. Para los enfermos con hirsutismo aislado es útil adminis trar el antangonista androgénico espironolactona.
Hirsutismo
Uno de los motivos más frecuentes de consulta endocrinológica es el exceso de vello corporal en las mujeres. Tres aspectos son de utilidad para decidir el tipo de evaluación que debe realizarse [ver tabla 1]: ¿Cuándo comenzó el exceso en el crecimiento de vello? ¿Existe un patrón semejante en otros familiares? ¿Las menstruaciones son regulares o irregulares?
El hirsutismo familiar que comienza en la pubertad y se acompaña de periodos regulares es un trastorno sin gran importancia, y la mayoría de las pacientes solo necesitan tranquilizarse al respecto. Se requiere poca investigación endócrina, pero puede intentarse el tratamiento casmético y la administración de espironolactona.
El hirsutismo que es desproporcionado a los antecedentes étnicos de la paciente y que se acompaña de menstruaciones normales se denomina idiopático. Aunque no suele ser necesario realizar estudios endócrinos, una investigación simple puede incluir la medición de la testos terona total y libre, y de la DHEAS. Si las concentraciones de testosterona y de DHEAS son normales, la paciente puede ser tranquilizada respecto a que el trastorno es benigno.
En ocasiones las pacientes con hirsutismo y menstruaciones normales tienen HSCAT. El inicio del hirsutismo en la pubertad con menstruaciones irregulares debe hacer pensar en síndrome de ovarios poliquísticos. El hiperandrogenismo ovárico es la alteración subyacente máscomún, que se refleja con frecuencia, aunque no en forma invariable, por elevación en la concentración de testosterona. Debido a que la HSCAT puede causar síndrome de ovarios poliquísticos, debe con siderarse la medición de la 17-OHP, en especial en una paciente que desea ser fértil.
Las pacientes con hirsutismo idiopático y sin alteraciones menstruales buscan reducir el crecimiento del pelo. Con frecuencia todo lo que se necesita son enfoques cosméticos, como el uso de ceras, depiladores o electrólisis. Las opciones endócrinas incluyen el uso de anticonceptivos orales, que tienen múltiples efectos, incluyendo la reducción de las concentraciones de testosterona libre en sangre y el antagonismo androgénico en el tejido blanco. El mejor tipo de anticonceptivo oral para usarse para disminuir el crecimiento del pelo es el que contenga un componente progestágeno no androgénico, como el Demulen. La administración del antagonista de la aldosterona, la espironolactona, es útil tanto sola como aunada a un anticonceptivo oral. Con frecuencia se requieren dosis de 100 a 150 mg por día. En los pacientes con diabetes mellitus o deterioro renal este medicamento puede inducir con rapidez hipercalemia severa, y debe evitarse. Por último, se ha usado el inhibidor de la Salfa-reductasa, el finasteride, con algunos resultados promisorios.105,106 Sin embargo, su uso aún no está autorizado en los Estados Unidos.
El hirsutismo de reciente aparición en una mujer adulta, en especial cuando se asocia con amenorrea, requiere una investigación completa para descartar un tumor suprarrenal u ovárico. Si existen signos de síndrome de Cuching deberá seguirse el protocolo diagnóstico ya mencionado. En estos casos debe medirse la DHEAS para detectar el exceso de andrógenos suprarrenales. Por otro lado, si existe virilización pura, debe medirse la testosterona en plasma y las concentraciones de DHEAS. Si la concentración de testosterona es de 1.5 ng/ml o mayor, deben obtenerse imágenes tanto de los ovarios como de las suprarrenales. Si está elevada la concentración de DHEAS debe considerarse la supresión suprarrenal con dexametasona y realizar una TC de estas glándulas.107,108
La médula suprarrenal
FISIOLOGIA
La médula suprarrenal se compone de tejido cromafín de origen neuroectodérmico. Es la fuente principal de epinefrina en el organismo, sintetizada a partir de la tirosina a través de un compuesto intermediario, la dopamina. La enzima dopamina b-hidroxilasa cataliza la hidroxilación de la dopamina a norepinefrina (que se sintetiza en menor proporción en la médula suprarrenal), y ésta a su vez es metilada para formar epinefrina por medio de la enzima catecolamina N-metiltransferasa. Esta enzima es estimulada por el cortisol que llega a la médula directa mente de la corteza suprarrenal. De esta manera la función cortical regula la función de la médula suprarrenal.
La epinefrina tiene numerosos efectos sobre el metabolismo intermedio, incluyendo la estimulación de la glucogenólisis hepática, la inhibición del agluconeogénesis hepática y la inhibición de la liberación de insulina; también estimula la liberación de ácidos grasos libres por medio de la lipólisis de triglicéridos en el tejido adiposo. La epinefrina tiene los siguientes efectos sobre el sistema cardiovascular: inotropismo cardiaco positivo, produce una respuesta vasopresora y ejerce un efecto combinado de vasoconstricción y vasodilatación en diferentes lechos vasculares. Resulta paradójico que la epinefrina constituya uno de los principales reguladores metabólicos, pero que no sea necesario sustituirla en los pacientes con suprarrenalectomía o enfermedad de Addison.109,110
FEOCROMOCITOMA
La única enfermedad importante relacionada con la médula suprarrenal es el feocromocitoma, tumor muy vascularizado y con gran capacidad secretora.111-113 Se considera que la hipertensión arterial intermitente es su característica principal, pero cerca del 15 porciento de los pacientes presentan hipertensión sostenida, el 45 porciento cursan con hipertensión paroxística y el 40 porciento con hipertensión sostenida más paroximos. Los síntomas relacionados que deben llamar la atención hacia el diagnóstico son: diaforesis, cefalea, cuadros de palpitaciones, hipermetabolismo, alteraciones mentales incluyendo psicosis e insuficiencia cardiaca congestiva secundaria a miocarditis o a necrosis miocárdica focal.114 La hipotensión ortostática es un hallazgo frecuente en el examen físico; en parte se debe a la disminución del volumen circulante. Muchas enfermedades poco frecuentes se pueden relacionar con el feocromocitoma; el carcinoma medular de tiroides es el más común y es importante diagnosticarlo pues es curable. Una discusión amplia sobre el diagnóstico y tratamiento del feocromocitoma aparece en la Sección I, Subsección VII.
Reconocimientos
Figura 1 Albert Miller.
Figura 2 Alan D. Iselin.
DR. DANIEL D. FEDERMAN
Fisiología de la corteza suprarrenal
La glándula suprarrenal controla los ajustes del organismo en la posición erecta y permite la adaptación a la ingestión intermitente de alimentos. En ocasiones se requieren también aumentos súbitos y sostenidos en la función suprarrenal para tolerar episodios agudos de estrés como la pérdida de volumen, la infección, la anestesia y la cirugía. La pérdida de estas capacidades constituye la consecuencia más importante de la insuficiencia suprarrenal. Las secreciones suprarrenales también ejercen una influencia sobre la respuesta inmune, la formación de células sanguíneas, la función cerebral, la síntesis de proteínas y sobre muchos otros procesos que se llevan a cabo en el organismo.1 Al exceso de estos efectos se le conoce como síndrome de Cushing.
BlOSlNTESlS Y METABOLISMO DE LOS ESTEROIDES SUPRARRENALES
La síntesis de las hormonas suprarrenales se inicia a partirdel acetato o del colesterol [ver figura 1]. El colesterol derivado de las lipoproteínas de baja densidad (LDL) parece ser el sustrato para la biosíntesis del 80 porciento de los esteroides. La conversión del colesterol a D5- pregnenolona es el punto principal de regulación por las hormonas tróficas que controlan la función suprarrenal; tanto la corticotropina como la angiotensina estimulan este paso. Después de la síntesis de pregnenolona las vías metabólicas divergen hacia la formación de cada uno de los productos biológicamente activos de la suprarrenal.
|
| Figura 1 |
| Biosíntesis de las hormonas suprarrenales |
Cortisol En los seres humanos el cortisol es el principal glucocorticoide. Influye sobre el apetito y el bienestar, tiene un papel primordial en el mantenimiento de los niveles de glucosa en la sangre al promover la gluconeogénesis hepática, y afecta en forma indirecta la frecuencia cardiaca y la función de bomba al controlar la síntesis de epinefrina en la médula suprarrenal. El aumento en la secreción de cortisol es una característica crucial en la respuesta fisiológica al estrés y a la enfermedad.
De 13 a 20 mg de cortisol son secretados cada día por las zonas fascicular y reticular; inmediatamente después de ser liberado por la glándula, el cortisol se une a la µ- globulina transcortina, o globulina fijadora de cortisol. Como en el caso de la tiroxina, la mayor parte del cortisol circulante se encuentra inactivo, unido a las proteínas; una pequeña proporción está libre y es biológicamente activo. La reducción, a nivel hepático, del doble enlace en el anillo A de la molécula inactiva al cortisol; después de varias biotransformaciones y conjugaciones con glucurónidos o sulfatos, los metabolitos del cortisol se excretan como 17 hidroxiesteroides (17-OHS) en la orina.
Andrógenos Los andrógenos se producen en la zona fascicular y reticular; su producción varía considerablemente en las distintas etapas de la vida. El feto elabora grandes cantidades de andrógenos suprarrenales, en especial de sulfato de dehidroepiandrosterona (DHEAS), mientras que el niño produce muy poco.2 La producción aumenta durante la pubertad y alcanza niveles máximos en los adultos jóvenes, que rápidamente comienzan a disminuir a niveles más bien bajos en las personas mayores de cincuenta años de edad. En cambio, la secreción de la hormona adrenocorticotrópica (ACTH), único mecanismo de control conocido de la biosíntesis de andrógenos suprarrenales, no muestra tales fluctuaciones relacionadas con la edad. La regulación de la producción de andrógenos suprarrenales no es muy clara: se han postulado factores tanto derivados de la hipófisis como de la suprarrenal, aunque no se han confirmado. Los andrógenos suprarrenales son relativamente poco potentes, pero sirven como precursores para la conversión hepática a testosterona [ver figura 2]. La hiperfunción de esta vía metabólica en las mujeres produce hirsutismo y, en ocaciones, virilización.
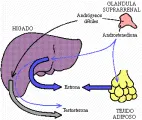
|
| Figura 2 |
| Síntesis de andrógenos |
La androstenediona, otro andrógeno suprarrenal, puede ser transformado por el hígado y el tejido adiposo a estrona, un estrógeno potente. De hecho, está es la principal fuente de estrógenos en niños y mujeres posmenopáusicas. El otro andrógeno importante, la dehidroepinadrosterona (DHEA), junto con su conjugado DHEAS, constituye un enigma. La DHEA tiene una potencia androgénica mínima, y aunque se han publicado algunas acciones no adrogénicas, ninguna parece tener importancia fisiológica demostrada. En forma semejante, la DHEAS puede metabolizarse a testos terona o a estrógeno, pero su importancia a este respecto no es clara aún. Los andrógenos suprarrenales son metabolizados a compuestos 17-ceto, que se excretan en la orina como 17-cetoesteroides (17-KS).
Aldosterona El principal mineralocorticoide producido por la corteza suprarrenal es la aldosterona, un 18 oxicorticoide producido en la capa glomerular a partir de sus precursores, acetato y colesterol.3 La angiotensina constituye el estímulo dominante para la biosíntesis de aldosterona [ver figura 3]; tanto la angiotensina como la ACTH aumentan la secreción de aldosterona al acelerar la conversión de colesterol a D5-pregnenolona. Otros estímulos para la secreción de aldosterona incluyen el aumento del potasio sérico, la pérdida de volumen sanguíneo, la disminución del sodio sérico y el aumento en los niveles de estrógenos. Existen evidencias de que la liberación de aldosterona es inhibida por mecanismos dopaminérgicos. La aldosterona circula libremente y es degradada enzimáticamente durante la depuración hepática, así como por conjugación y excreción renal. Su grado de depuración metabólica depende estrechamente del aporte sanguíneo hepático. La disminución de éste, como ocurre en diversos trastornos hepáticos, puede producir hiperaldosteronismo transitorio; las interacciones con otros mecanismos que controlan la secreción de aldosterona determinan si esta alteración perdura.
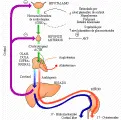
|
| Figura 3 |
| Control de la función suprarrenal |
REGULACION DE LA SECRECION SUPRARRENAL
La ACTH es el principal regulador de la producción de cortisol y andrógenos suprarrenales [ver figura 3].3 A su vez, la síntesis y liberación de ACTH por la hipófisis es controlada sobre todo por la hormona liberadora de corticotropina (CRH, por sus siglas en inglés, n. el t.), un péptido de 41 aminoácidos producido en el hipotálamo.4
La vasopresina, también llamada arginina vasopresina (AVP) u hormona antidiurética (HAD), también participa en la regulación basal de la ACTH.5,6 Aunque la vasopresina, la oxitocina y las catecolaminas influyen en el ciclo diurno de ACTH y cortisol, el principal determinante de este ciclo parece ser la liberación diurna de CRH. Durante los estados tensionales aumenta de dos a 10 veces la producción de esteroides suprarrenales; esta respuesta está en relación con un efecto potencial, tanto de la CRH como de la HAD, más que un simple efecto aditivo.
La ACTH se sintetiza como un pequeño segmento de la compleja molécula llamada pro-opiomelanocortina (POMC).l0 El producto primario del genes una molécula compleja que también incluye las secuencias de aminoácidos para la b-lipoproteína (b-LPH), un péptido de unión, las hormonas estimuladoras de los melanocitos µ y b (µ-MSH y b-MSH), la b-endorfina, la µ-LPH, la metencefalina y al menos dos fragmentos adicionales, el péptido del lóbulo intermedio parecido a la corticotropina (CLIP, por sus siglas en inglés, n. del t.) y una secuencia de 16 kilobases (16 kb) de significado fisiológico no determinado [ver figura 4]. La POMC también se produce en tejidos no hipofisarios, pero su procesamiento postraducción difiere en varios aspectos. En la hipófisis una hendidura separa al fragmento más largo al principio de la ACTH y ambas porciones son glucosiladas; más tarde, los fragmentos más pequeños son liberados. Aunque la liberación de la b-lipoproteína tiende a ser paralela a la de ACTH, la función de esta molécula y la de los otros dos fragmentos del precursor mayor no está completamente aclarada.
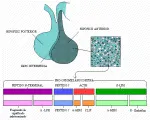
|
| Figura 4 |
| Síntesis de ACTH |
Al igual que otras hormonas hipofisiarias, la ACTH se secreta en pulsos.7 Ocurren de uno a tres pulsos cada hora, con una concentración mayor por la mañana. Debido a que el cortisol resultante desaparece más lentamente que la ACTH, los niveles matutinos iniciales se suman al ritmo diurno, que es mayor durante la mañana y menor por la tarde. Además de la producción de pulsos endógenos, la liberación de ACTH y cortisol también es estimulada por la fiebre, la hipoglucemia, las tensiones de todo tipo, los trastornos psicológicos y la disminución en los niveles de cortisol plasmático. El factor dominante en la inhibición de ACTH es el nivel plasmático de cortisol. Todos los glucocorticoides sintéticos tienen el mismo efecto, que es mediado por la supresión de la transcripción de ARN mensajero (ARNm) para la síntesis de POMC.8 Se piensa que una supresión similar por los glucocorticoides inhibe la producción de CRH. Por lo tanto, el cortisol tiene un efecto de retroalimentación negativa a nivel tanto hipota lámico como hipofisiario [ver figura 3].
Pruebas de función suprarrenal
Las principales pruebas utilizadas para evaluar la función suprarrenal son la determinación en plasma de ACTH y cortisol y de 17-OHS urinarios y cortisol libre en orina de 24 horas.
Determinación de cortisol y ACTH en plasma Los valores basales de cortisol plasmático fluctúan entre 10 y 25 µg/100 ml por la mañana y 2 y 10 µg/ml durante la noche. El nivel de cortisol en plasma varía por los cambios en la cantidad de globulina fijadora de cortisol, que suelen ser paralelos a los de la globulina fijadora de tiroxina. Por ejemplo, ambas proteínas se elevan por el estímulo estrogénico y disminuyen en las enfermedades hepáticas. Los niveles basales de cortisol suelen ser menos importantes que su respuesta a los estímulos fisiológicos; por lo tanto, los niveles de cortisol plasmático habitualmente se miden después de la administración de un esteroide exógeno para determinar si el eje hipófisis-suprarrenal es susceptible a la inhibición por retroalimentación, es decir, si puede suprimirse, y después de la administración de ACTH para observar si la suprarrenal responde a la estimulación.
A continuación se describen algunas pruebas bien estandarizadas para evaluar la función suprarrenal:
1. Para determinar la supresión del eje hipófisis suprarrenal, se administra 1 mg de dexametasona (un análogo del cortisol) por vía oral a las 11 :P.M., y se cuantifican los niveles de cortisol plasmático a las 8:00 A.M. del día siguiente. Los valores normales son menores de 5 µg/100 ml.
2. Para determinar la respuesta suprarrenal, se administran por vía intravenosa 0.25mg de cosintropina (ACTH sintética). Se mide el cortisol plasmático a los 0 y 60 minutos; un valor de 20 µg/dl en cualquier momento de la prueba indica respuesta suprarrenal normal.
3. Para evaluar la reserva hipófisis-suprarrenal, se administran 3 g de metirapona por vía oral a media noche, y se determinan los niveles plasmáticos de cortisol y desoxicortisol (compuesto S) a las 8:00 A.M. del día siguiente. La metirapona inhibe la 11-hidroxilación, lo que ocasiona disminución en los niveles de cortisol y aumento de ACTH. Si la reserva hipófisis-suprarrenal está intacta, los niveles de cortisol en plasma deben ser menores de 5 µg/100 ml y los de desoxicortisol mayores de 10 µg/100 ml.
4. Un método alternativo para evaluar la reserva hipófisissuprarrenal consiste en la administración intravenosa de insulina, 0.15U/kg. La hipoglucemia secundaria debe provocar respuestas tanto de cortisol (normal, 20 µg/dl) como de hormona del crecimiento (> 8 ng/ml). Esta prueba debe realizarse en presencia de un médico preparado para administrar 25 ml de glucosa al 50 porciento por vía intravenosa en caso de una reacción de hipoglucemia.
5. Para determinar la reserva hipofisiaria de ACTH se puede administrar CRH ovina en dosis de 1 µg/kg de peso corporal. Tanto los niveles de ACTH como los de cortisol en plasma se elevan en los individuos normales. Esta prueba puede realizarse a cualquier hora del día, pero su sensibilidad es mayor por la tarde.
La ACTH en plasma puede determinarse por prueba de inmunorradiometría (IRMA), que emplea anticuerpos contra las porciones N-terminal y C-terminal de la hormona. A diferencia de la prueba de radioinmunoensayo, la IRMA puede distinguir en forma confiable los niveles bajos, normales y elevados de ACTH.9
Niveles de 17-hidroxiesteroides urinarios Aunque tiene la ventaja de integrar la secreción de cortisol durante un periodo de 24 horas, la utilidad de la determinación de 17-OHS urinarios es limitada, ya que éstos se elevan en la obesidad y disminuyen en las enfermedades hepáticas y renales. Por otra parte, la recolección adecuada de la muestra urinaria es difícil para muchos pacientes.
Cortisol libre en orina de 24 horas Esta prueba también requiere que la recolección de la muestra se realice de forma adecuada. Los valores en individuos obesos rara vez se superponen con los de aquellos pacientes con enfermedad de Cushing, y los trastornos hepáticos no alteran los resultados. La excreción urinaria de cortisol libre se ha convertido en un valioso indicador de hiperfunción suprarrenal. Es menos seguro para detectar insuficiencia suprarrenal.l0
Hiperfunción de la corteza suprarrenal: Síndrome de Cushing
El síndrome de Cushing es un estado poco frecuente de hiperfunción de la corteza suprarrenal. Sin embargo, es importante clínicamente por dos razones. En primer lugar, porque es a menudo curable. En segundo lugar, porque el exceso de glucocorticoides es una causa potencial de los problemas que el médico observa cotidianamente, como obesidad, hipertensión arterial, diabetes mellitus, litiasls renal, osteoporosis, púrpura, trastornos mentales, alteraciones menstruales, impotencia, hirsutismo, edema, hipocalemia, susceptibilidad a las infecciones y cicatrización deficiente. Aunque ninguno de estos poblemas hace pensar per se en el diagnóstico de hipercortisolismo, la coexistencia de varias características clínicas pueden justificar la investigación de síndrome de Cushing. Entre las más útiles se encuentran la facies pletórica, las equimosis, la presencia de hipocalemia de causa desconocida, los defectos en la cicatrización y los cambios en las facciones, basándose en la comparación del aspecto actual con una fotografía previa.1l
Las causas del síndrome de Cushing son el exceso de ACTH, el aumento en la producción de cortisol por las suprarrenales y la ingestión iatrogénica o facticia de corticoides.l2
EXCESO DE ACTH
Producción hipofisaria excesiva (Enfermedad de Cushing)
El exceso de ACTH hipofisaria con hiperplasia suprarrenal bilateral es la causa de casi dos tercios de los casos de síndrome de Cushing. Esta enfermedad es más frecuente en mujeres en edad reproductiva y su aparición suele ser paulatina. Cualquier síntoma producido por el exceso de cortisol puede aparecer inicialmente, pero finalmente ocurren trastornos menstruales en las mujeres, impotencia en los hombres, aparición de equimosis, facies de luna llena y plétora generalizada. La secreción de andrógenos suprarrenales está aumentada, por lo que en las mujeres son frecuentes el hirsutismo y la alopecia leve. El exceso de andrógenos puede ejercer cierto efecto protector contra la influencia antianabólica del cortisol, pero la gravedad de las manifestaciones clínicas es más o menos proporcional a la duración del trastorno y a la cantidad de ACTH secretada. La pigmentación, una manifestación del exceso de ACTH, puede ser un dato de gran valor para el diagnóstico, aunque este signo puede ser difícil de reconocer, sobre todo en individuos de piel oscura.
En teoría, puede producirse un exceso de ACTH hipofisaria por un aumento de la hormona liberadora de corticotropina o por una neoplasia primaria de la hipófisis. Cushing consideraba que la mayoría de los casos eran causados por un tumor hipofisario y en la actualidad la enorme experiencia de curación secundaria a la extirpación de microadenomas de hipófisis apoya esa interpretación. Los estudios moleculares han indicado que la mayoría de los microadenomas productores de ACTH son monoclonales.13 Como en el caso de otros tumores, esto es una fuerte evidencia de que el origen de la neoplasia es hipofisiario, más que una respuesta hiperplásica a CRH. Sin embargo, la recurrencia de los tumores después de una aparente mejoría y la respuesta de algunos pacientes a fármacos como la bromocriptina, agonista de la dopamina, apoyan, aunque no demuestran, un origen hipotalámico. Por el contrario, el éxito de la radioterapia hipofisaria, especialmente en niños, apoya la opinión de que, como afirmó Cushing, la enfermedad de Cushing es con más frecuencia un hiperadrenalismo secundario a tumor hipofisario.
Los siguientes datos son característicos de la enfermedad de Cushing:
1. Elevación de los niveles de cortisol libre urinario, por lo general mayores de 150 µg/24 horas.
2. Pérdida del ritmo circadiano de la ACTH y el cortisol plasmáticos: los niveles vespertinos de cortisol son mayores de 10 µg/dl; la excreción urinaria de 17-OHS también pierde su ritmo diario.
3. Pérdida de la capacidad para suprimir la secreción de ACTH con dosis fisiológicas de glucocorticoides: la administración de 1 mg de dexametasona a media noche no impide la elevación matutina de los niveles de cortisol por encima de 10 µg/dl.
4. Inhibición de la secreción de cortisol cuando se administran dosis mayores de dexametasona, 2 mg cada seis horas durante dos días: los niveles urinarios de 17-OHS y 17-KS disminuyen a menos del 50 porciento de las cifras control.
Producción ectópica de ACTH
Existen dos variantes de síndrome de Cushing causado por la producción ectópica de ACTH (no hipofisiaria). La primera es el resultado de la producción intensa de ACTH por neoplasias malignas, siendo las más frecuentes las de pulmón, timo, páncreas y riñón.14 El síndrome consiste en hipercorticismo agudo, en el que predominan los efectos mineralocorticoides secundarios a la secreción de grandes cantidades de cortisol, más que los cambios inducidos por su acción glucocorticoide. La alcalosis hipocalémica, la debilidad y la pérdida de peso son los elementos clínicos sobresalientes y el aspecto cushinoide es poco frecuente. El cortisol plasmático y los esteroides urinarios se encuentran a menudo mucho más elevados y el paciente rara vez tiene una franca apariencia cushinoide. Además, los niveles urinarios o plasmáticos de 17-OHS no se suprimen por una dosis diaria de 8 mg de dexametasona. Por tanto, el dato principal para establecer el diagnóstico de este tipo de producción ectópica de ACTH es la discrepancia entre un cuadro clínico poco llamativo y la presencia de niveles muy elevados en la secreción de esteroides. El gran efecto mineralocorticoide en este padecimiento es consecuencia de la gran producción de cortisol y de la saturación de las dos vías de inactivación del mismo (la conversión acortisona por la 11-hidroxiesteroide deshidrogenasa y la reducción del anillo A a dehidrocortisol). Como consecuencia se acumula cortisol, que se une al receptor de los mineralocorticoides y causa retención de sodio con caliuresis.15,16
La segunda forma de síndrome de Cushing ectópico se relaciona con lesiones benignas o malignas como carcinoides bronquiales o tímicos. El cuadro clínico en estos casos es indistinguible del síndrome de Cushing secundario a alteraciones hipofisarias y son necesarios estudios especiales para hacer la diferenciación [ver adelante, Diagnóstico].
Producción ectópica de CRH
La producción ectópica de CRH por tumores puede estimular una producción suficiente de ACTH en la hipófisis para ocasionar hiperplasia suprarrenal y síndrome de Cushing. El tumor más implicado en esta producción ectópica de CRH es el carcinoide bronquial, y la mayor parte de estos tumores secretan también ACTH.1 El cuadro clínico puede ser típico de enfermedad de Cushing o presentar las manifestaciones del síndrome de secreción ectópica de ACTH como hipocalemia, alcalosis y debilidad.17 La prueba con dexametasona en dosis altas y bajas no muestra inhibición de la función hipofisiaria-suprarrenal, y los niveles de ACTH en plasma se encuentran muy elevados.
EXCESO DE CORTISOL SUPRARRENAL
Las neoplasias suprarrenales hipersecretoras de cortisol constituyen menos de la tercera parte de los casos de síndrome de Cushing espontáneo. Aunque los síntomas varían, hay ciertos datos clínicos que son útiles para el diagnóstico. Los adenomas suprarrenales benignos a menudo secretan sólo cortisol, por lo que las manifestaciones clínicas reflejan el exceso de cortisol sin aumento de andrógenos. Debido a que el cortisol suprime la secreción endógena de ACTH, otras funciones suprarrenales se en cuentran disminuidas y los 17-cetosteroides tienden a ser bajos. Los carcinomas suprarrenales que producen cortisol en exceso casi siempre producen grandes cantidades de andrógenos; en la mujer aparece hirsutismo y virilización y los cetosteroides urinarios tienden a ser muy altos (50 mg/ 24 horas).l8 Otras características diagnósticas que indican la presencia de tumores suprarrenales incluyen una amplia variabilidad en la actividad secretora, la falta de supresión por altas dosis de dexametasona, alteraciones en la urografía excretora (UE) en el lado afectado, y una masa palpable en cerca del 50 por ciento de los casos de carcinoma suprarrenal.
La menos frecuente y más enigmática causa de síndrome de Cushing es la hiperplasia nodular bilateral no dependendiente de ACTH. Se han postulado varios mecanismos raros, incluyendo mutaciones activadoras de los receptores de proteína G que estimulan a la adenilato ciclasa.19 La hiperplasia macronodular es muy probable que se presente como una complicación tardía de la enfermedad de Cushing no tratada, en la cual uno o más nódulos se hacen autónomos. Dependiendo de los niveles de producción autónoma del cortisol, la ACTH hipofisaria puede estar elevada o aparentemente normal; las pruebas con dosis altas de dexametasona deben inhibir a la ACTH pero no el cortisol. Por lo menos en un caso se comprobó la progresión de dependencia hipofisaria a la autonomía suprarrenal.20
La causa más confusa de síndrome de Cushing es la displasia suprarrenal micronodular bilateral, trastorno que en ocasiones es de tipo familiar y en el que los niveles de cortisol están elevados y los de ACTH bajos, observándose en las suprarrenales nódulos pequeños activos y áreas de tejido atrófico.21 En algunas familias la hiperplasia suprarrenal parece deberse a una inmunoglobulina estimuladora de las suprarrenales, un autoanticuerpo hacia los receptores de ACTH similar a las inmunoglobulinas estimuladoras del tiroides en la enfermedad de Graves.
La causa menos frecuente de hiperplasia suprarrenal nodular es el hipercortisolismo dependiente de alimentos, en el que la expresión inadecuada del receptor del polipéptido inhibitorio gástrico (PIG) por las células suprarrenales provoca una secreción exagerada de cortisol en respuesta a los alimentos (pero no en la mañana). Este tipo recién conocido de síndrome de Cushing podría ser muy importante y pueden implicarse mecanismos iguales o parecidos en otros trastornos.22-24
Más adelante se discute el exceso de cortisol por ingestión iatrogénica o facticia [ver adelante, Uso farmacológico de los glucocorticoides].
DIAGNOSTICO
Por las diversas manifestaciones del exceso de los glucocorticoides, es evidente la utilidad de contar con una prueba simple y sencilla que excluya el diagnóstico de síndrome de Cushing en un paciente con pocas probabilidades de tenerlo. Para este fin la prueba más adecuada es la cuantificación de cortisol plasmático matutino después de administrar 1 mg de dexametasona la noche anterior. Si el cortisol se encuentra por debajo de 5µg/dl, se descarta el síndrome de Cushing. Si los valores se encuentran por encima de esta cifra y el paciente no está recibiendo tratamiento estrogénico, está indicada la evaluación completa y definitiva para establecer el diagnóstico. Esta prueba no confirma el diagnóstico en un paciente con probabilidad de síndrome de Cushing. Debido a la gran prevalencia de obesidad cushinoide (quizá 3,000 casos por millón en adultos), la ausencia de supresión por la dexametasona en estos pacientes tiene muy poco significado a menos que la evidencia clínica de síndrome de Cushing sea muy fuerte.25
Cuando se sospecha clínicamente el síndrome de Cushing, se puede confirmar mediante la prueba con 1 mg de dexametasona o la determinación del cortisol libre urinario en 24 horas. Debido a que la prueba en plasma se asocia con más falsas positivas, se usa más la determinación de cortisol en orina. Cuando existen dudas acerca del diagnóstico se recomienda realizar ambas pruebas.
Si la prueba de escrutinio fue positiva, la evaluación definitiva debe incluir lo siguiente [ver figura 5]:

|
| Figura 5 |
| Enfoque diagnóstico del síndrome de Cushing |
l. Administración de dexametasona, 0.5 mg cada seis horas durante dos días. Los niveles de l7-OHS en orina de 24 horas menores de 3.5 mg en el segundo día o cifras de cortisol plasmático inferiores a 5 µg/dl en la mañana del tercer día descartan el síndrome de Cushing; los niveles mayores lo confirman. Debe enfatizarse la importancia de que este punto quede totalmente claro. Aunque parezca irónico, las pruebas para localizar la causa patológica del síndrome de Cushing no son útiles para establecer su presencia. Por lo tanto, debe demostrarse sin duda alguna que existe hipercortisolismo antes de realizar más estudios.
2. El primer paso para establecer la causa consiste en distinguir entre el hipercortisolismo dependiente de ACTH y el independiente. Para ello deben realizarse varias mediciones plasmáticas de ACTH [ver figura 6] con determinación simultánea de cortisol. Los nuevos estudios inmunorradiométricos para ACTH permiten diferenciar con claridad los valores bajos y suprimibles de los tumores adrenales (< 5 pg/ml) de los valores aparentemente normales o elevados de los casos dependientes de ACTH.26 Un nomograma elaborado en la Clínica Mayo puede identificar el origen patológico del hipercortisolismo en alrededor del 90 porciento de los casos midiendo el cortisol en plasma, el cortisol libre en orina y los niveles de ACTH.27

|
| Figura 6 |
| Diagnóstico diferencial del síndrome de Cushing |
3. Administración de dexametasona, 2 mg cada seis horas durante dos días. Los siguientes resultados son característicos de exceso de ACTH hipofisiaria (enfermedad de Cushing): un nivel de 17-OHS en orina el segundo día menor del 35 porciento del basal, un nivel de cortisol libre urinario el segundo día menor del 10 porciento del basal o una concentración de cortisol en plasma la tercera mañana menor de 10 µg/dl.28 El fracaso para llegar a estos resultados indica una fuente no hipofisiaria de ACTH o un tumor o hiperplasia suprarrenales autónomos. El valor de la ACTH determinada en el inciso 2, mencionado antes, permitirá esta distinción. La prueba con dosis altas de dexametasona se ha modificado para evitar la incomodidad e inexactitud de la recolección urinaria. Se obtiene una determinación de la concentración basal de cortisol a las 8 A.M., se administran 8 mg de dexametasona por vía oral a las 11 P.M. y se obtiene una segunda medición de cortisol a las 8 A.M. del día siguiente. Un valor de menos del 50 por ciento del basal el segundo día indica origen hipo fisiario,10 pero la sensibilidad y especificidad de esta prueba para hipercortisolismo dependiente de ACTH es mayor si se combina con la prueba estándar de supresión con dosis altas (8 mg/día).29
4. Si los niveles de ACTH son normales o elevados, la tomografía computada (TC) o la resonancia magnética (IRM) de la hipófisis pueden indicar la presencia de un tumor. Si la ACTH está inhibida, se deberá realizar una TC o IRM del área suprarrenal.
5 Los pasos anteriores consiguen un diagnóstico definitivo en el 90 porciento de los pacientes. Los resultados endócrinos y de imágenes se combinan para establecer un origen hipofisiario o ectópico de la ACTH o un tumor suprarrenal como la causa del hipercortisolismo. El problema más difícil de solucionar consiste en distinguir entre la producción de ACTH hipofisaria excesiva y la producción ectópica por alguna variedad de carcinoide [ver antes, Producción excesiva de ACTH].30 Si no se detecta un tumor hipofisario se recomienda realizar una TC del tórax, ya que el 90 porciento de los tumores benignos productores de ACTH y CRH se localizan en esa área.
6.Para los pacientes ocasionales en quienes la distinción de la patología sigue siendo un problema, pueden utilizarse dos estudios recientes. La inyección por vía intravenosa de hormona liberadora de corticotropina ovina (CRHo), 100 µg o l µg/kg provoca una elevación del ACTH y del cortisol plasmático en la enfermedad de Cushing, pero no en el hipercorticismo independiente de ACTH. Sin embargo, todavía persiste cierta sobreposición, en particular con algunos pacientes con producción ectópica de ACTH que responden a esta prueba.31-34
7. La prueba más sofisticada y difícil desde el punto de vista técnico para identificar microadenomas demasiado pequeños para ser visualizados es la administración simultánea de CRHo (i.e., estimulación hipofisaria) y la toma de muestras de ambos senos petrosos inferiores.35 La concentración dos veces mayor de ACTH entre las venas petrosas y las venas periféricas sugiere un adenoma hipofisario y justifica la exploración transesfenoidal. La concentración dos veces mayor de ACTH entre ambas venas petrosas y las cifras basales o después de la administración de CRH se considera una justificación para realizar hemihipofisectomía si no se identifica quirúrgicamente un microadenoma.
Un enfoque nuevo y elegante para localizar producción ectópica de ACTH consiste en el uso de ocreótido radiomarcado. Este análogo de larga acción de la somatostatina se fija a los receptores expresados en los tumores neuroendócrinos como los carcinoides bronquiales, pero no a los adenomas hipofisiarios habituales. Por lo tanto, puede usarse para diagnosticar, localizar y tratar la producción ectópica de ACTH.36,37
Cuando se realizan y correlacionan cuidadosamente, las pruebas descritas deben proporcionar la interpretación definitiva en casi todos los casos de síndrome de Cushing [ver figura 6]. Sin embargo, es indispensable empezar por comprobar que el paciente tiene hipercortisolismo. A menos que el cortisol libre en orina esté elevado, es probable que el examen diferencial y de localización sea confusa e inútil.
Es conveniente considerar algunos aspectos útiles respecto a la evaluación del eje hipófisis-suprarrenal. Algunas circunstancias pueden alterar el ritmo circadiano de la secreción de ACTH y cortisol, produciendo resistencia a la supresión hipofisaria con 1 mg de dexametasona. La más evidente es el estado tensional agudo, como el provocado por la hospitalización o una intervención quirúrgica; los niveles vespertinos de cortisol después de los primeros días de estancia hospitalaria pueden llegar a estar por encima de 10 µg/dl sin acompañarse de datos clínicos anormales. Una segunda causa de dificultades es la depresión y agitación que pueden simular alteraciones en las pruebas dinámicas compatibles con síndrome de Cushing por exceso de secreción de ACTH hipofisaria. En pacientes con depresión ansiosa se pierde el ritmo circadiano y la capacidad de supresión con dexametasona. Esta situación constituye un problema especial porque el hipercorticismo a su vez puede causar depresión ansiosa. El diagnóstico diferencial se confirma por el examen clínico para detectar la presencia de signos del síndrome, y cuantificando las cifras de cortisol libre urinario, que es normal en el deprimido. En el paciente deprimido con nivel urinario de cortisol elevado puede ser útil la prueba de CRH.38
Las dos enfermedades más difíciles de diferenciar del síndrome de Cushing son la obesidad y el alcoholismo; en estas tres afecciones se puede encontrar plenitud facial, debilidad, fatiga y equimosis. En ocasiones los resultados de las pruebas de escrutinio en individuos obesos son anormales, pero las pruebas dinámicas finalmente permiten distinguir a estos sujetos de los que tienen síndrome de Cushing. En el seudosíndrome de Cushing que ocurre en los alcohólicos, las pruebas iniciales pueden rnostrar resistencia a la supresión del cortisol plasmático con la administración de 1 mg de dexametasona y pérdida del ritmo circadiano. Sin embargo, con la abstinencia los valores de cortisol plasmático vuelven a la normalidad y reaparece la capacidad de supresión con dexametasona.39
El hipercortisolismo cíclico o periódico es otra causa de hallazgos de laboratorio confusos e inquietantes en el síndrome de Cushing. Los ciclos pueden variar entre días a meses y pueden persistir durante años. Se requiere de gran cuidado antes de recomendar el tratamiento quirúrgico.40
TRATAMIENTO
El tratamiento de elección en adultos con síndrome de Cushing es la cirugía, y en la forma más común del mismo, la enfermedad de Cushing (i.e., exceso de la producción hipofisaria de ACTH), la micro adenectomía transesfenoidal es el tratamiento de elección.41-45 Si las imágenes de la hipófisis (la IRM es la técnica de mayor sensibilidad) o la prueba del seno petroso identifican un microadenoma [ver figura 7], las cifras de curación son tan altas como del 90 porciento. Ocurren recurrencias en el 5 al 20 porciento de los casos.46 Se obtienen mejores resultados cuando la silla turca es radiológicamente normal. Cuando hay alteraciones en la silla turca o el adenoma es mayor de 10 mm de diámetro, las oportunidades de curación son claramente menores. Se han observado complicaciones como diabetes insípida transitoria y meningitis, pero son fáciles de tratar.
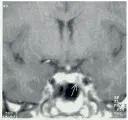
|
| Figura 7 |
| Adenoma hipofisiario |
Cuando la microadenectomía tiene éxito los pacientes muestran niveles plasmáticos de cortisol normales o bajos pronto después de la cirugía; dichos pacientes suelen presentar supresión de la función hipofisaria y suprarrenal y pueden necesitar tratamiento sustitutivo durante periodos tan largos como seis meses después de la cirugía.47 Cuando los hallazgos quirúrgicos son poco definidos o cuando los niveles de cortisol no bajan, se puede administrar una inyección de CRH para demostrar la persistencia de células productoras de ACTH. Otras técnicas incluyen tratamiento con partículas pesadas, implante de itrio y radiaciones externas. Todos estos tratamientos son inferiores a la adenomectomía, aunque es importante señalar que, en la mayoría de las instituciones, la radiación externa es el tratamiento de elección en niños.
El entusiasmo actual por el enfoque directo a la hipófisis no debe menospreciar la utilidad ocasional de la adrenalectomía bilateral. En aquellos pacientes con enfermedad grave, complicaciones que ponen en peligro la vida, tumores hipofisarios muy grandes o hiperplasia nodular y en los que el tratamiento hipofisario ha fracasado, la suprarrenalectomía puede ser preferible a una microadenectomía transesfenoidal.48 El tratamiento preoperatorio con metirapone puede modular el hipercortisolismo, incluyendo las alteraciones psicológicas, lo que hace que la cirugía sea más segura. Todos los pacientes manifiestan insuficiencia suprarrenal después de la suprarrenalectomía y un porcentaje importante desarrollan síndrome de Nelson. El autotransplante de tejido suprarrenal se ha utilizado con el fin de evitar la insuficiencia suprarrenal, pero pocas veces se ha logrado implantar una cantidad de tejido suprarrenal que evite la hipofunción y que no ocasione recurrencia del trastorno.
Existen varios tratamientos farmacológicos para el síndrome de Cushing. La aminoglutetimida inhibe la síntesis de cortisol al bloquear la conversión del coleterol a D5- pregnenolona. El mitotane es un agente adrenolítico que inhibe la síntesis de cortisol, y el metirapone reduce la producción de cortisol al inhibir la 11-hidroxilasa. Sin embargo, la toxicidad por la aminogutetimida y el mitotane es alta; incluso cuando ambos medicamentos se administran en combinación para disminuir su toxicidad individual, los resultados rara vez son satisfactorios. Por lo tanto, la utilidad de los fármacos en el síndrome de Cushing se limita a situaciones especiales, como el control de manifestaciones severas antes de lacirugía, como complemento de la remisión parcial inducida por irradiación hipofisaria, o para aliviar los síntomas en enfermos con carcinoma metastásico inoperable o con producción ectópica de ACTH.
La resección quirúrgica es el tratamiento de elección para el carcinoma o el adenoma suprarrenal. Si la extirpación del tumor tiene éxito, se presenta insuficiencia suprarrenal y el paciente necesita tratamiento con corticoesteroides en forma temporal. La recuperación de la función normal hipotalámica, hipofisaria y suprarrenal puede retrasarse por periodos tan prolongados como dos años.49
Síndrome de Nelson
Del 10 al 40 porciento de los pacientes con enfermedad de Cushing (i.e., exceso de ACTH hipofisaria) que son tratados con suprarrenalectomía bilateral desarrollan síndrome de Nelson.50 Este síndrome, caracterizado por hiperpigmentación notable y un tumor cromófobo de la hipófisis, puede desarrollarse entre seis meses y I5 años después de la suprarrenalectomía. En algunos casos el crecimiento local agresivo de la tumoración causa oftalmoplejía, defectos visuales y, en ocasiones, infarto hipofisario. Debido a que se desconoce qué características del síndrome de Cushing anteceden al desarrollo del síndrome de Nelson, los pacientes tratados con suprarrenalectomía bilateral deben vigilarse a intervalos regulares. El diagnóstico se confirma mediante la demostración de hiperpigmentación, agrandamiento de la silla turca y niveles elevados de ACTH.
Las concentraciones de ACTH en plasma después de la adrenalectomía para el síndrome de Cushing tienden a ser mayores a las observadas en la enfermedad espontánea de Addison y con frecuencia aumentan 20 veces lo normal, hasta 300 a 1,000 pg/ml. Es probable, aunque no seguro, que la determinación anual de los niveles de ACTH permitan detectar la enfermedad antes de que se manifieste clínicamente. El tratamiento incluye resección quirúrgica, radioterapia con partículas pesadas, ciproheptadina y bromocriptina. Los informes difieren con respecto a si la irradiación profiláctica de la hipófisis evita el desarrollo del síndrome de Nelson posadrenalectomía.
Uso farmacológico de los glucocorticoides
Parael clínico, los glucocorticoides han sido fármacos de gran valor que a la vez le han ocasionado serios problemas. Aunque su uso en ciertas enfermedades llega a salvar la vida, producen síndrome de Cushing en algunos pacientes y supresión del eje hipotálamo-hipófisis-suprarrenal en todos los enfermos sometidos a tratamiento crónico.5l Ciertas complicaciones son características del sindrome de Cushing iatrogénico, incluyendo hipertensión intracraneana benigna, glaucoma, cataratas y pancreatitis.5l
Aunque rara vez se ha demostrado que la supresión del eje hipotálamo-hipófisis-suprarrenal cause la muerte, sigue siendo preocupante en los sujetos tratados con glucocorticoides. Es imposible determinar el tiempo mínimo de tratamiento necesario para que se establezca la supresión de la función endógena; si el tiempo lo permite, puede realizarse la prueba corta con ACTH. Se administra una dosis de 0.25 mg de cosintropin por vía intravenosa y, si el cortisol en plasma a los 30 minutos es mayor de 18 ng/dl, el eje hipotálamo-hipófisis se considera normal. Sin embargo, con frecuencia no hay tiempo para esperar el resultado, por lo que es conveniente administrar suplementos de esteroides a cualquier paciente tratado antes con estos fármacos. La revisión de la historia y la experiencia con la insuficiencia suprarrenal secundaria han sugerido que los suplementos farmacológicos con esteroides son excesivos y que la dosis debe depender de dos aspectos: la dosis recibida por tiempo prolongado y la magnitud de la enfermedad o cirugía. En casos de cirugía menor es adecuado administrar 25 a 50 mg/día de hidrocortisona o su equivalente, para situaciones intermedias 50 a 75 mg/día y para cirugía mayor 100 a 150 mg/día durante uno a tres días.52 Esta dosis puede disminuirse en un 50 porciento cada día, comenzando dos días después de la cirugía si todo va bien.
La recuperación total de la supresión intensa del eje hipotálamo-hipófisis-suprarrenal puede tardar hasta dos años después de la suspensión del tratamiento, y no parece existir manera de acelerar este proceso. Durante este periodo los pacientes pueden manifestar síntomas de supresión a pesar de tener niveles basales normales de cortisol, así como de cierta respuesta de la hipófisis ante el estímulo. Para valorar la recuperación, la mejor prueba parece ser la respuesta del cortisol plasmático ante el estímulo con ACTH o a la hipoglucemia inducida por insulina.53 El nivel basal o estimulado mayor de 18 µg/dl indica una respuesta suprarrenal normal. Sin embargo, ninguna prueba es con fiable al 100 porciento. La prueba de estimulación con ACTH sintética puede en ocasiones ser normal en pacientes que responden en forma subnormal al estrés.54
Aldosteronismo primario
El aldosteronismo primario es un síndrome manifestado por hipertensión arterial e hipocalemia que ocurre en el 0.05 a dos porciento de los pacientes hipertensos55. Entre las causas predominan los adenomas unilaterales (en alrededor del 70 porciento de los casos), la hiperplasia bilateral idiopática (en el 20 a 30 porciento) y, en raras ocasiones, un carcinoma adrenocortical.56-58 Los pacientes con aldosteronismo primario se encuentran entre la población hipertensa que tiene una concentración limítrofe de potasio, en el rango de 3.4 a 3.6 mEq/L, o de hipocalemia franca que ocurre en forma espontánea o por el uso de diuréticos leves. En los pacientes que reciben diuréticos éstos deben ser suspendidos durante cuatro semanas y se repletará la deficiencia de potasio. A continuación se administra al paciente una dieta que proporcione por lo menos 100 mEq de sal al día y se miden la aldosterona y la renina en plasma a las 8 A.M. después de que el paciente ha estado recostado toda la noche y de nuevo cuando el paciente ha estado de pie durante cuatro horas. También se mide la concentración de aldosterona en orina de 24 horas. Además de la hipocalemia causada por la carga de sodio, en los pacientes con aldosteronismo primario la concentración plasmática de renina es menor de 1 ng/ml/h, la de aldosterona es mayor de 15 ng/dl y los metabolitos urinarios de la aldosterona estan elevados. Después de localiza el tumor o se confirma la hiperplasia por medio de tomografía computada y por medición de aldosterona y cortisol en ambas venas suprarrenales.59
La hipertensión y la hipocalemia del síndrome de Conn (adenoma unilateral) suelen responder a la suprarrenalectomíaunilateral. Los pacientes con hiperplasia se tratan médicamente. Se ha sugerido que la obtención de una sola relación de actividad plasmática de aldosterona/renina puede usarse como método de escrutinio para el hiperaldosteronismo primario.60,61
La hipertensión e hipocalemia inducidas por la ingestión de orozuz es secundaria al acúmulo de ácido glicirrízico en las células tubulares renales, con la consecuente inhibición de una enzima (la 11 13-hidroxiesteroide deshidrogenasa) que inactiva los mineralocorticoides.16 El aldosteronismo remediable por glucocorticoides (ARG) típicamente se manifiesta en la infancia, y existe gran frecuencia de hipertensión severa con eventos cerebrovasculares entre los adultos jóvenes de las familias afectadas. Como en el síndrome de Conn, la hipocalemia es común, pero no es suficientemente frecuente como para ser útil para el escrutinio. El ARG es un trastorno autosómico dominante causa do por la formación mutante de un gen quimérico en la zona fasciculada cuyo producto proteico combina las activida des de la 11b-hidroxilasa y la sintetasa de aldosterona. Este defecto causa exceso de aldosterona que depende de ACTH en lugar de angiotensina. Al suprimir la ACTH los glucocorticoides disminuyen tanto la hipertensión como la hipocalemia, Como en otros trastornos autosómico dominantes, el escrutinio ha descubierto mucho más individuos afectados de lo que antes de sospechaba.62
Hipofunción de la corteza suprarrenal
La insuficiencia suprarrenal aguda es un trastorno poco frecuente que suele desarrollarse durante una enfermedad grave. La causa más cornún es la hemorragia suprarrenal por anticoagulantes, y parece existir una mayor asociación con la trombocitopenia inducida por heparina y con el síndrome de anticuerpos antifosfolípido.63,64 La insuficiencia también puede presentarse en pacientes con sepsis aguda o después de cirugía, infarto del miocardio o embarazo.65 Los datos clínicos incluyen náusea, debilidad, hipotensión arterial, colapso y dolor abdominal o en la espalda. Este dolor puede ser el único dato que indica la complicación.66-68
El diagnóstico es sencillo, las pruebas sistemáticas de laboratorio probablemente muestren niveles bajos de sodio sérico y niveles elevados de potasio y BUN. Sin embargo, la evaluación definitiva requiere cuantificar los niveles de cortisol plasmático antes y 60 minutos después de la administración de 0.25 mg de ACTH sintética (Cortrosyn). Si al diagnóstico se retrasa excesivamente, se deben administrar 100 mg de hidrocortisona por vía intravenosa al paciente seguidos de 10 mg de cortisol por hora hasta que se obtengan los resultados de las pruebas. En situaciones graves la prueba de ACTH debe diferirse y se debe empezar el tratamiento de inmediato, después de haber tomado la muestra para la determinación del cortisol plasmático. La concentración de cortisol menor de 10 µg/dl cuando la muestra se examina en retrospectiva sugiere por lo menos insuficiencia suprarrenal parcial, un valor por arriba de 18 µLg/dl indica una respuesta normal al estrés. La TC y la IRM pueden confirmar el diagnóstico de hemorragia suprarrenal.
ENFERMEDAD DE ADDISON
La insuficiencia suprarrenal crónica, o enfermedad de Addison tiene dos causas principales. En la actualidad las infecciones granulomatosas crónicas, como la tuberculosis o la histoplasmosis, son la causa de una minoría de los casos en los Estados Unidos. La causa más frecuente es un trastorno autoinmune en el que la invasión suprarrenal por linfocitos y células plasmáticas se acompaña de anticuer pos antisuprarrenales en el plasma. Esta entidad puede aparecer sola o con hipotiroidismo secundario a tiroiditis de Hashimoto [ver adelante, Síndrome de deficiencia poliendócrina]. La insuficiencia suprarrenal puede ocurrir también como manifestación de trastornos hereditarios caracterizados por la degeneración progresiva de la mielina, ya sea en el cerebro (adrenoleucodistrofia) o en la médula espinal (adrenomielodistrofia).
La insuficiencia suprarrenal puede ser una complicación del síndrome de inmunodeficiencia adquirida (SIDA).69 Las suprarrenales se infiltran con patógenos oportunistas y por sarcoma de Kaposi. Aunque se ha observado disminución de la reserva suprarrenal en varias series de pacientes, la insuficiencia suprarrenal típica, confirmada por pruebas y por la respuesta al tratamiento, se ha visto con poca frecuencia en pacientes con SIDA. El tratamiento de pacientes con SIDA con megestrol, un análogo de progestinas con posible efecto anabólico, suprime en ocasiones el eje hipófisis-suprarrenal y causa hipoadrenalismo.70-72
La tomografía computada ha permitido aclarar la etiología de la insuficiencia suprarrenal. Las suprrarrenales aumentadas de tamaño sugieren adrenalitis tuberculosa activa73 o tumor metastásico, cada uno con importantes implicaciones clínicas. Los pacientes en los que se sospeche tuberculosis activa deben recibir tratamiento definitivo. Ha surgido una consecuencia interesante de esta decisión: el tratamiento con rifampicina puede precipitar insu ficiencia suprarrenal en los pacientes con Addison tratados o no tratados.74 Este estado de insuficiencia ocurre porque el medicamento induce enzimas hepáticas que metabolizan esteroides y que causan la depuración acelarada de la secreción endógena subnormal o de las cantidades terapéuticas habituales de hormonas suprarrenales. El anestésico etomidato y el antibiótico antimicótico ketoconazol pueden también inducir insuficiencia suprarrenal reversible, pero su efecto es secundario a un deterioro en la biosíntesis de esteroides.77,76 Los pacientes con metástasis en las suprarrenales pueden presentar insuficiencia súbita, por lo que cuando se encuentra crecimiento de estas glándulas en pacientes con enfermedad metastásica conocida debe considerarse la administración profiláctica de remplazo esteroideo.77
Los pacientes con insuficiencia suprarrenal crónica presentan debilidad, anorexia, pérdida de peso, hipotensión e hipovolemia. Un dato útil para el diagnóstico es la presencia de hiperpigmentación sobre los pliegues de las palmas de las manos y otras áreas corporales, en puntos de presión y alrededor de las aréolas y pezones. Excepto por la tendencia a la hipercalemia, los resultados de los exámenes de laboratorio de rutina no son característicos hasta que ocurre el colapso suprarrenal, caracterizado por hiponatremia, hipercalemia, hipoglucemia, hemoconcentración y elevación del nitrógeno ureico. Las alteraciones en el nitrógeno ureico y los electrolitos son secundarios en gran parte a la deficiencia de volumen y a la azoemia prerrenal. La única prueba diagnóstica especi fica es la determinación de los niveles de cortisol plasmático o de los 17-OHS, antes y después de la estimulación con ACTH . La selección de la prueba diagnóstica dependerá de la valoración del estado clínico del paciente.
Si no parece existir insuficiencia suprarrenal y es necesaria alguna prueba para descartar esta posibilidad, la inyección intravenosa de ACTH sintética, en dosis de 0.25 mg, es sensible y específica. Se determinael nivel plasmático de cortisol antes de la inyección y 60 minutos después. La respuesta normal consiste en un aumento de al menos 18 µg/dl o mayor [ver figura 8]. Sin embargo, si el valor basal es mayor de 20 µg/dl, el aumento puede ser mínimo y la función suprarrenal debe considerarse normal.

|
| Figura 8 |
| Diagnóstico de la insuficiencia suprarrenal |
En aquellos individuos cuyo cuadro clínico es compatible con insuficiencia suprarrenal (i.e., que tienen hiponatremia o hipotensión), el estudio es un poco diferente. Se realiza una medición matutina de cortisol y ACTH y después, para evitar el colapso suprarrenal, el paciente puede recibir 0.5 mg de dexametasona, y fludrocortisona, 0.1 mg al día, durante la prueba. Se mide el cortisol a los 30 y 60 minutos después de la administración de 0.25 mg de ACTH sintética. La concentración de cortisol en plasma por arriba de 18 µg/dl establece una función suprarrenal normal; sin embargo, si la medición inicial muestra que el cortisol es bajo y sigue bajo con una ACTH por arriba de 100 pg/ml, se establece el diagnóstico de insuficiencia suprarrenal primaria.78 Los enfermos con insuficiencia suprarrenal secundaria a hipopituitarismo pueden diferenciarse de otros sujetos con insuficiencia suprarrenal primaria, o enfermedad de Addison por la presencia de deficiencia de otras hormonas hipofisarias y por un nivel de ACTH , en el límite inferior normal o por debajo del mismo. La insuficiencia suprarrenal secundaria puede confirmarse también administrando metirapone (30 mg/kg por vía oral administrado con los alimentos a media noche). Un nivel en plasma de ll-desoxicortisol por arriba de 7 µg/dl confirma la función normal del eje hipotálamo-hipófisis suprarrenal (HHS). Esa prueba debe hacerse con el pacien te internado por la posibilidad de inducir insuficiencia suprarrenal. En ocasiones la prueba de metirapone descubre una respuesta subnormal en un paciente con prueba normal de ACTH aguda.79 En estos pacientes, en especial en los que tienen menos de seis semanas desde el inicio de la deficiencia suprarrenal, la prueba con ACTH intravenosa puede ser normal porque no ha ocurrido aún atrofia suprarrenal.
En los pacientes con sospecha de crisis suprarrenal no debe permitirse ningún retraso para realizar procedimientos diagnósticos; se toma una muestra de sangre para la determinación del cortisol plasmático y a través de la misma aguja se administra la primera dosis de tratamiento. Es mejor administrar el tratamiento sustitutivo para la insuficiencia suprarrenal aguda con hemisuccinato de hidrocortisona por vía intravenosa, un bolo inicial de 100 mg seguido de 10 mg/hr durante los dos primeros días de la crisis. Para restaurar el volumen circulante es necesario utilizar soluciones mixtas (glucosadas y salinas), expansores del plasma, y en algunos casos sangre total. Debido a que el paciente con enfermedad de Addison suele tener deficiencias considerables de volumen antes de desarrollar la crisis, puede ser necesaria la administración de grandes volúmenes de líquido. Si el estado del individuo mejora puede reducirse la dosis de esteroides de forma gradual, hasta los niveles de mantenimiento, durante tres a cinco días.
El tratamiento sustitutivo para la insuficiencia suprarrenal crónica es de 20 a 25 mg de cortisona al levantarse por la mañana y de 10 a 15 mg por la tarde. Los pacientes también deben recibir fludrocortisona, 0.05 a 0.10 mg por vía oral cada día; los efectos de la dosis deben vigilarse, incluyendo el aumento de peso o la aparición de hipocalemia. La dosis de esteroides deberá incrementarse a discreción, siempre que cualquier individuo con disfunción suprarrenal crónica se exponga a un estado tensional importante. Para una infección de vías respiratorias superiores o influenza es suficiente con duplicar la dosis diaria de cortisona. Sin embargo, si un paciente con enfermedad de Addison sufre vómito, es aconsejable hospitalizarlo para administrar líquidos por vía intravenosa e hidrocortisona.
INSUFICIENCIA SUPRARRENAL SECUNDARIA
Los pacientes con hipopituitarismo que incluye deficiencia de ACTH sufren un síndrome semejante a la enfermedad de Addison excepto porque tienen menor grado de deficiencia de volumen. Debido a que el principal mecanismo de control de la aldosterona es la angiotensina, la regulación del volumen y electrolitos está menos afectada en individuos con panhipopituitarismo que en pacientes con enfermedad de Addison. Los enfermos con deficiencia de cortisol de cualquier causa, incluyendo el pahipopituitarismo, son propensos a la sobrecarga de líquidos y es probable que desarrollen hiponatremia. De manera característica, los niveles de ACTH en plasma están elevados en sujetos con insuficiencia suprarrenal primaria y son normales o no detectables en aquellos con deficiencia de ACTH primaria.
La CRH, actualmente disponible, puede ayudar a diferenciar entre las causas hipotalámicas e hipofisiarias de la insuficiencia suprarrenal secundaria. Si la prueba de intolerancia a la insulina no estimula la producción de cortisol a la hipoglucemia, existe un hipoadrenalismo secundario. Si en dicho paciente la prueba de CRH causa una rápida liberación de ACTH, se sospecha una probable enfermedad hipotalámica; si la CRH no provoca la liberación de ACTH, se supone que la lesión está en la hipófisis.80
SINDROMES POR DEFICIENCIA DE ALDOSTERONA
En ocasiones la deficiencia de aldosterona es la manifes tación única o predominante en la insuficiencia suprarrenal. Los defectos aislados en la biosíntesis de aldosterona son extremadamente raros,81 pero los defectos parciales forman parte del cuadro de hiperplasia suprarrenal congénita secundaria a la deficiencia de 21-hidroxilasa. Los síntomas atribuibles a la deficiencia de aldosterona algunas veces proporcionan los datos iniciales principales para el diagnóstico de insuficiencia suprarrenal; por ejemplo, puede presentarse hipotensión postural, antes que anorexia, pérdida de peso, astenia e hiperpigmentación.
Dos síndromes por deficiencia aislada de aldosterona merecen mención aparte, el hipoaldosteronismo idiopático y el hipoaldosteronismo hiporreninémico.82 El primero es el hipoaldosteronismo idiopático, trastorno muy raro que se presenta como un bloqueo cardiaco secundario a hipercalemia, o como una hipotensión ortostática secundaria a hipovolemia con o sin hiponatremia importante. Sería de esperar que en estos pacientes los niveles plasmáticos y urinarios de aldosterona se encuentren elevados, Junto con un aumento en la actividad de renina plasmática, aunque existen pocos casos publicados con estas características. Para el tratamiento del hipoaldosteronismo idiopático resulta eficaz la fludrocortisona, en dosis de 0.05 a 0.10 mg por día, combinada con la ingestión libre de sal.
El hipoaldosteronismo hiporreninémico es mucho más frecuente que el hipoaldosteronismo idiopático, y en sus formas leves es probable que el diagnóstico pase inadvertido. Los pacientes típicos son mayores de 45 años y padecen insuficiencia renal crónica. El trastorno renal afecta más al tejido tubular e intesticial que a los glomérulos; la diabetes mellitus es una causa frecuente. La característica principal del hipoaldosteronismo hiporreninémico es la hipercalemia crónica, de leve a severa, que en ocasiones se exacerba súbitamente por una hiperglucemia. También suele presentarse acidosis metabólica hiperclorémica, con niveles de sodio normales o bajos. La restricción de la ingesta de sodio exacerba las manifestaciones clínicas. La alteración fisiopatológica de fondo consiste en una concentración baja de renina en plasma a pesar de existir hipercalemia, contracción del volumen circulante e hiponatremia. Aunque la causa del bajo nivel de renina está poco clara, suele atribuirse a un defecto en el aparato yuxtaglomerular. Sin embargo, se ha demostrado que la deficiencia de prostaglandinas inducida por antinflamatorios no esteroides (AINE) puede ser una causa reversible del síndrome; en otros casos, el trastorno se agrava por la administración de heparina, bloqueadores de los canales del calcio o por bloqueadores beta adrenérgicos. El tratamiento del hipoaldosteronismo hiporreninémico incluye la corrección de la acidosis, ingestión libre de sodio y administración cuidadosa de fludrocortisona a las dosis necesarias para controlar la elevación de potasio sérico sin provocar insuficiencia cardiaca congestiva . La heparina es un inhibidor potente de la producción de aldosterona con natriuresis consecuente e hiponatremia ocasional. Ocurre hipercalemia en menos del 10 porciento de los pacientes, pero que puede ser rápida y severa en los enfermos con insuficiencia renal o diabetes mellitus o en enfermos que toman medicamentos que elevan la concentración de potasio sérico.83
Síndromes de deficiencia poliendócrina
Así como la hiperfunción y, las neoplasias pueden afectar a más de una glándula , de la misma forma puede ocurrir con las deficiencias endócrinas. El término síndrome de deficiencia poliendócrina (o síndromes autoinmunes poliglandulares) se refiere a dos tipos de deficiencia endócrina que se superponen, pero que tienen algunas características distintivas.84-86 El tipo I incluye hipoparatiroidismo, hipoadrenalismo y moniliasis mucocutánea, con anemia perniciosa y hepatitis crónica como manifestaciones ocasionales; el inicio en la niñez es característico. El tipo II, también llamado síndrome de Schmidt, incluye insuficiencia suprarrenal, tiroiditis de Hashimoto y, con menor frecuencia, diabetes mellitus insulinodependiente; la enfermedad se inicia a menudo alrededor de los 30 años de edad. Los siguientes hechos plantean una etiología autoinmune: la infiltración linfocítica de las glándulas, el hallazgo frecuente de anticuerpos antiglandulares y la asociación con trastornos autoinmunes conocidos, como la anemia perniciosa y la alopecia. En algunos pacientes ocurre insuficiencia gonadal primaria prematura. Los pacientes con estos síndromes poliglandulares deben vigilarse estrechamente, ya que pueden desarrollar una segunda o tercera alteración endócrina. Así mismo, dado que los síndromes de deficiencia poliendócrina son trastornos familiares, con patrón de herencia de tipo autosómico recesivo,87 es necesario someter a estudio a otros miembros de la familia para descartar la hipofunción endócrina.
Neoplasias suprarrenales e incidentalomas
Una minoría de las neoplasias suprarrenales tienen actividad endócrina.88 Tanto los adenomas como los carcinomas pueden producir síndrome de Cushing y síndromes de virilización. Los carcinomas son típicamente ineficaces en su patrón biosintético. Debido a que los carcinomas suprarrenales pueden sintetizar tipos o cantidades inapropiadas de esteroides, los valores de esteroides urinarios pueden proporcionar la clave para el diagnóstico. Por ejemplo, debe sospecharse cáncer suprarrenal en aquellos pacientes con síndrome de Cushing que presentan una concentración desproporcionadamente alta de 17-KS urinarios en relación con los 17-OHS, o en mujeres con datos de virilización que excretan más de 50 mg de 17-KS al día. Aunque la virilización es poco frecuente en los adenomas benignos, se han publicado varios casos que sólo producían testosterona. Estos casos justifican una mención especial,89-92 porque los niveles urinarios de 17-KS no se encontraban elevados, como suele suceder en las neoplasias suprarrenales que producen hirsutismo, y porque dos de los tumores respondieron a la administración de gonadotropina coriónica aumentando su producción de testosterona. En varios aspectos estos tumores, aunque de origen suprarrenal, se comportan como si se derivaran del tejido gonadal. Las neoplasias feminizantes son las tumoraciones suprarrenales menos frecuentes; deben de considerarse en el diagnóstico de varones con signos de feminización, sobre todo si presentan cifras elevadas de 17-KS, así como de estrógenos93,94
El amplio uso de la TC de abdomen ha dado lugar a un nuevo problema: la detección casual de una masa suprarrenal. Si la lesión es un quiste o un mielolipoma, no se requieren más estudios. Si la lesión es compatible con una masa sólida solitaria, se recomienda un procedimiento de escrutinio económico para detectar un tumor productor de hormonas, Algunos autores investigan solo hiperaldosteronismo y feocromocitoma; sin embargo, el autor considera que el estudio debe incluir la prueba de supresión con 1 mg de dexametasona para el síndrome de Cushing, la determinación de potasio sérico para el aldosteronismo, la medición del sulfato de dehidroepiandrosterona (DHEAS) en plasma o de 17-KS en orina de 24 horas para detectar exceso de andrógenos, y la determinación de ácido vanililmandélico (VMA) en orina de 24 horas para descartar feocromocitoma. La prueba de supresión con dexametasona desenmascara en ocasiones la disfunción en pacientes que no tienen un nivel suprimido de cortisol ni tampoco apariencia cushinoide.95-97 Estos pacientes pueden presentar insuficiencia suprarrenal si se extirpa el adenoma.93 Si se confirma alguna alteración endócrina, está indicada la cirugía. Para las lesiones no funcionantes el tamaño de la lesión es una guía útil.
Las lesiones de menos de 6 cm de diámetro casi siempre son benignas, mientras que las mayores se asocian con tal frecuencia de malignidad que justifica su extirpación.
La aspiración con aguja fina (después de que se ha extirpado el feocromocitoma) puede ser útil. Si se identifica un tumor metastásico puede buscarse el tumor primario. Si se detecta un tumor suprarrenal, aunque la citología no sea concluyente, la combinación de la apariencia más el tamaño de la lesión permitirá una decisión apropiada en relación con la cirugía.99-101
Hiperplasia suprarrenal congénita
La hiperplasia suprarrenal congénita (HSC) engloba un grupo de trastornos caracterizados por deficiencias enzimáticas hereditarias en la biosíntesis del cortisol; estos defectos tienen como consecuencia aumentos en los niveles de corticotropina, que a su vez producen exceso de andrógenos. En el sexo femenino, el hiperandrogenismo produce masculinización de los genitales externos en la etapa fetal y, si éste no es tratado, ocasiona amenorrea primaria, hipoestrinismo e hirsutismo en la pubertad. En más del 90 porciento de los casos el defecto se encuentra en la enzima 21 -hidroxilasa; con menor frecuencia se afectan la 11-hidroxilasa, la 3-1b-ol-deshidrogenasa, la 17 hidroxilasa y otras enzimas que participan en la síntesis del cortisol. El tratamiento con cortisol produce supresión en la secreción de ACTH, disminuye de los niveles plasmáticos elevados de los precursores de cortisol y corrige las manifestaciones clínicas.
El gen de la deficiencia de la enzima 21-hidroxilasa, junto con un seudogen relacionado, se localiza en el cromosoma 6 dentro del segmento HLA. Varias mutaciones distintas de estos genes han sido identificadas como causa de la deficiencia enzimática que provoca la HSC.102 La incidencia en todo el mundo de deficiencia de 21-hidroxilasa es de alrededor de uno por 14,000 nacimientos, pero existe importante heterogeneidad genética y no todas las manifestaciones son fáciles de explicar.
La deficiencia completa de 21-hidroxilasa suele manifestarse en el recién nacido con virilización e hipertrofia de los genitales. Las expresiones más leves de este padecimiento tienen relevancia clínica en la adolescencia y juventud. Los individuos heterocigotos y homocigotos para el defecto (identificados por estudios genéticos del fenotipo HLA) presentan grados variables de hirsutismo y trastornos menstruales. Por lo tanto, algunos pacientes que presentan hirsutismo, oligomenorrea o incluso síndrome de ovarios poliquísticos idiopáticos manifiestan exceso de andrógenos suprarrenales secundario a defectos parciales en la biosíntesis del cortisol. En los casos en que no hay una virilización congénita el síndrome se denomina hiperplasia suprarrenal congénita de aparición tardía (HSCAT). El método diagnóstico inicial más simple consiste en determinar la 17-OH progesterona (I7-OHP) antes de las 9:00 A.M. durante la fase folicular del ciclo menstrual. Si el nivel de este precursor del colesterol es de 200 ng/dl o menor, el diagnóstico se excluye. Si es de 500 ng/dl o mayor se establece el diagnóstico. Los pacientes con nive les entre estos rangos deben recibir 0.25 mg de ACTH sintética por vía intravenosa y medirse la 17-OHP a los 60 minutos. Los invidividuos afectados tienen concentraciones de 17-OHP mayores de 1,500 ng/dl. Los pacientes con valoresentre l,000 y 1,500 ng/dl pueden ser heterocigotos.103 Existe importante desacuerdo sobre la frecuencia de esta alteración y sobre la prueba adecuada para demostrar las expresiones leves de la enfermedad.104
Una consecuencia práctica de un resultado positivo es que la supresión parcial del eje hipófisis-suprarrenal, como la inducida por la administración de 5 mg de prednisona en el momento de acostarse, puede mejorar la irregularidad menstrual y disminuir el hirsutismo. Las deficiencias par ciales de la 11-hidroxilación y de la 3b-hidroxiesteroide deshidrogenasa también han sido identificadas como posibles variantes de HCSAT que producen hirsutismo. La supresión con prednisona, como se describió antes, es adecuada para tratar a los pacientes con estas deficiencias. Para los enfermos con hirsutismo aislado es útil adminis trar el antangonista androgénico espironolactona.
Hirsutismo
Uno de los motivos más frecuentes de consulta endocrinológica es el exceso de vello corporal en las mujeres. Tres aspectos son de utilidad para decidir el tipo de evaluación que debe realizarse [ver tabla 1]: ¿Cuándo comenzó el exceso en el crecimiento de vello? ¿Existe un patrón semejante en otros familiares? ¿Las menstruaciones son regulares o irregulares?
|
||||||||||||||||||||||||
|
El hirsutismo familiar que comienza en la pubertad y se acompaña de periodos regulares es un trastorno sin gran importancia, y la mayoría de las pacientes solo necesitan tranquilizarse al respecto. Se requiere poca investigación endócrina, pero puede intentarse el tratamiento casmético y la administración de espironolactona.
El hirsutismo que es desproporcionado a los antecedentes étnicos de la paciente y que se acompaña de menstruaciones normales se denomina idiopático. Aunque no suele ser necesario realizar estudios endócrinos, una investigación simple puede incluir la medición de la testos terona total y libre, y de la DHEAS. Si las concentraciones de testosterona y de DHEAS son normales, la paciente puede ser tranquilizada respecto a que el trastorno es benigno.
En ocasiones las pacientes con hirsutismo y menstruaciones normales tienen HSCAT. El inicio del hirsutismo en la pubertad con menstruaciones irregulares debe hacer pensar en síndrome de ovarios poliquísticos. El hiperandrogenismo ovárico es la alteración subyacente máscomún, que se refleja con frecuencia, aunque no en forma invariable, por elevación en la concentración de testosterona. Debido a que la HSCAT puede causar síndrome de ovarios poliquísticos, debe con siderarse la medición de la 17-OHP, en especial en una paciente que desea ser fértil.
Las pacientes con hirsutismo idiopático y sin alteraciones menstruales buscan reducir el crecimiento del pelo. Con frecuencia todo lo que se necesita son enfoques cosméticos, como el uso de ceras, depiladores o electrólisis. Las opciones endócrinas incluyen el uso de anticonceptivos orales, que tienen múltiples efectos, incluyendo la reducción de las concentraciones de testosterona libre en sangre y el antagonismo androgénico en el tejido blanco. El mejor tipo de anticonceptivo oral para usarse para disminuir el crecimiento del pelo es el que contenga un componente progestágeno no androgénico, como el Demulen. La administración del antagonista de la aldosterona, la espironolactona, es útil tanto sola como aunada a un anticonceptivo oral. Con frecuencia se requieren dosis de 100 a 150 mg por día. En los pacientes con diabetes mellitus o deterioro renal este medicamento puede inducir con rapidez hipercalemia severa, y debe evitarse. Por último, se ha usado el inhibidor de la Salfa-reductasa, el finasteride, con algunos resultados promisorios.105,106 Sin embargo, su uso aún no está autorizado en los Estados Unidos.
El hirsutismo de reciente aparición en una mujer adulta, en especial cuando se asocia con amenorrea, requiere una investigación completa para descartar un tumor suprarrenal u ovárico. Si existen signos de síndrome de Cuching deberá seguirse el protocolo diagnóstico ya mencionado. En estos casos debe medirse la DHEAS para detectar el exceso de andrógenos suprarrenales. Por otro lado, si existe virilización pura, debe medirse la testosterona en plasma y las concentraciones de DHEAS. Si la concentración de testosterona es de 1.5 ng/ml o mayor, deben obtenerse imágenes tanto de los ovarios como de las suprarrenales. Si está elevada la concentración de DHEAS debe considerarse la supresión suprarrenal con dexametasona y realizar una TC de estas glándulas.107,108
La médula suprarrenal
FISIOLOGIA
La médula suprarrenal se compone de tejido cromafín de origen neuroectodérmico. Es la fuente principal de epinefrina en el organismo, sintetizada a partir de la tirosina a través de un compuesto intermediario, la dopamina. La enzima dopamina b-hidroxilasa cataliza la hidroxilación de la dopamina a norepinefrina (que se sintetiza en menor proporción en la médula suprarrenal), y ésta a su vez es metilada para formar epinefrina por medio de la enzima catecolamina N-metiltransferasa. Esta enzima es estimulada por el cortisol que llega a la médula directa mente de la corteza suprarrenal. De esta manera la función cortical regula la función de la médula suprarrenal.
La epinefrina tiene numerosos efectos sobre el metabolismo intermedio, incluyendo la estimulación de la glucogenólisis hepática, la inhibición del agluconeogénesis hepática y la inhibición de la liberación de insulina; también estimula la liberación de ácidos grasos libres por medio de la lipólisis de triglicéridos en el tejido adiposo. La epinefrina tiene los siguientes efectos sobre el sistema cardiovascular: inotropismo cardiaco positivo, produce una respuesta vasopresora y ejerce un efecto combinado de vasoconstricción y vasodilatación en diferentes lechos vasculares. Resulta paradójico que la epinefrina constituya uno de los principales reguladores metabólicos, pero que no sea necesario sustituirla en los pacientes con suprarrenalectomía o enfermedad de Addison.109,110
FEOCROMOCITOMA
La única enfermedad importante relacionada con la médula suprarrenal es el feocromocitoma, tumor muy vascularizado y con gran capacidad secretora.111-113 Se considera que la hipertensión arterial intermitente es su característica principal, pero cerca del 15 porciento de los pacientes presentan hipertensión sostenida, el 45 porciento cursan con hipertensión paroxística y el 40 porciento con hipertensión sostenida más paroximos. Los síntomas relacionados que deben llamar la atención hacia el diagnóstico son: diaforesis, cefalea, cuadros de palpitaciones, hipermetabolismo, alteraciones mentales incluyendo psicosis e insuficiencia cardiaca congestiva secundaria a miocarditis o a necrosis miocárdica focal.114 La hipotensión ortostática es un hallazgo frecuente en el examen físico; en parte se debe a la disminución del volumen circulante. Muchas enfermedades poco frecuentes se pueden relacionar con el feocromocitoma; el carcinoma medular de tiroides es el más común y es importante diagnosticarlo pues es curable. Una discusión amplia sobre el diagnóstico y tratamiento del feocromocitoma aparece en la Sección I, Subsección VII.
Figura 1 Albert Miller.
Figura 2 Alan D. Iselin.
Bibliografía
- Orth DN, Kovaks WJ, DeBold CR: The adrenal cortex. William's Textbook of Endocrinology, 8th ed. Wilson JD, Foster DW, Eds. WB Saunders Co, Philadelphia, 1992, p 489
- Meikle AW, Daynes RA, Araneo BA: Adrenal androgen secretion and biologic effects. Endocrinol Metab Clin North Am 20:381, 1991 [PMID 1831756]
- Quinn SJ, Williams GH: Regulation of aldosterone secretion. Ann Rev Physiol 50:409, 1988
- Orth DN: Corticotropin-releasing hormone in humans. Endocr Rev 13:164, 1992
- Watabe T, Tanaka K, Kumagae M, et al: Role of endogenous arginine vasopressin in potentiating corticotropin-releasing hormone-stimulated corticotropin secretion in man. J Clin Endocrinol Metab 66:1132, 1988 [PMID 2836468]
- Salata RA, Jarrett DB, Verbalis JG, et al: Vasopressin stimulation of adrenocorticotropin hormone (ACTH) in humans. J Clin Invest 81:766, 1988 [PMID 2830315]
- Iranmanesh A, Lizarralde G, Short D, et al: Intensive venous sampling paradigms disclose high frequency adrenocorticotropin release episodes in normal men. J Clin Endocrinol Metab 71:1276, 1990
- Dallman MF, Akana SF, Levin N, et al: Corticosteroids and the control of function in the hypothalamo-pituitary-adrenal (HPA) axis. Ann NY Acad Sci 746: 22, 1994 [PMID 7825879]
- Findling JW, Engeland WC, Raff H: The use of immunoradiometric assay for the measurement of ACTH in human plasma. Trends in Endocrinol Metab 1:283, 1990
- Kaye TB, Crapo L: The Cushing syndrome: an update on diagnostic tests. Ann Intern Med 112:434, 1990
- Kannan CR: Diseases of the adrenal cortex. DM 34:613, 1988
- Sheeler LR: Cushing's syndrome. Cleve Clin J Med 55:329, 1988
- Biller BMK, Alexander JM, Zervas NT, et al: Clonal origins of adrenocorticotropin-secreting pituitary tissue in Cushing's disease. J Clin Endocrinol Metab 75:1303, 1992
- Odell WD: Ectopic ACTH secretion: a misnomer. Endocrinol Metab Clin North Am 20:371, 1991
- Ulick S, Wang JZ, Blumenfeld JD, et al: Cortisol inactivation overload: a mechanism of mineralocorticoid hypertension in the ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab 74:963, 1992 [PMID 1569172]
- Williams GH: Guardian of the gate: receptors, enzymes, and mineralocorticoid function (editorial). J Clin Endocrinol Metab 74:961, 1992
- Belsky JL, Cuello B, Swanson LW, et al: Cushing's syndrome due to ectopic production of corticotropin-releasing factor. J Clin Endocrinol Metab 60:496, 1985 [PMID 2982899]
- King DR, Lack EE: Adrenal cortical carcinoma: a clinical and pathologic study of 49 cases. Cancer 44:239, 1979 [PMID 455249]
- Malchoff CD, MacGillivray D, Malchoff DM: Adrenocorticotropic hormone-independent adrenal hyperplasia. Endocrinologist 6:79, 1996
- Hermus AR, Pieters GF, Smals AG, et al: Transition from pituitary-dependent to adrenal-dependent Cushing's syndrome. N Engl J Med 318:966, 1988 [PMID 2832758]
- Young WF Jr, Carney JA, Musa BU, et al: Familial Cushing's syndrome due to primary pigmented nodular adrenocortical disease: reinvestigation 50 years later. N Engl J Med 321:1659, 1989
- Reznik Y, Allali-Zerah V, Chayvialle JA, et al: Food-dependent Cushing's syndrome mediated by aberrant adrenal sensitivity to gastric inhibitory polypeptide. N Engl J Med 327:981, 1992 [PMID 1325609]
- Lacroix A, Bolte E, Tremblay J, et al: Gastric inhibitory polypeptide-dependent cortisol hypersecretion-a new cause of Cushing's syndrome. N Engl J Med 327:974, 1992
- Bertagna X: New causes of Cushing's syndrome (editorial). N Engl J Med 327: 1024, 1992
- Kreisberg R: Half a loaf. N Engl J Med 330:1295, 1994
- Findling JW: Clinical application of a new immunoradiometric assay for ACTH. Endocrinologist 2:360, 1992
- Snow K: Biochemical evaluation of adrenal dysfunction: the laboratory perspective. Mayo Clin Proc 67:1055, 1992
- Flack MR, Oldfield EH, Cutler GB Jr, et al: Urine free cortisol in the high-dose dexamethasone suppression test for the differential diagnosis of the Cushing syndrome. Ann Intern Med 116:211, 1992 [PMID 1728204]
- Dichek HL, Nieman LK, Oldfield EH, et al: A comparison of the standard high dose dexamethasone suppression test and the overnight 8-mg dexamethasone suppression test for the differential diagnosis of adrenocorticotropin-dependent Cushing's syndrome. J Clin Endocrinol Metab 78:418, 1994 [PMID 8106630]
- Zarate A, Kovaks K, Flores M, et al: ACTH and CRF-producing bron chial carcinoid associated with Cushing's syndrome. Clin Endocrinol 24:523, 1986
- Grossman AB, Howlett TA, Perry L, et al: CRF in the differential diagnosis of Cushing's syndrome: a comparison with the dexamethasone suppression test. Clin Endocrinol 29:167, 1988
- Nieman LK, Cutler GB Jr, Oldfield EH, et al: The ovine corticotropin-releasing hormone (CRH) stimulation test is superior to the human CRH stimulation test for the diagnosis of Cushing's disease. J Clin Endocrinol Metab 69:165, 1989 [PMID 2543689]
- Schulte HM, Allolio B, Günther TK, et al: Bilateral and simultaneous sinus petrosus inferior catheterization in patients with Cushing's syndrome: plasma-immunoreactive-ACTH-concentrations before and after administration of CRF. Horm Metab Res (suppl) 16:66, 1987
- Loriaux DL, Nieman L: Corticotropin-releasing hormone testing in pituitary disease. Endocrinol Metab Clin North Am 20:363, 1991 [PMID 1652435]
- Malchoff CD, Orth DN, Abboud C, et al: Ectopic ACTH syndrome caused by a bronchial carcinoid tumor responsive to dexamethasone, metyrapone, and corticotropin-releasing factor. Am J Med 84:760, 1988 [PMID 2840823]
- Lamberts SWJ, Reubi J-C: A role of (labeled) somatostatin analogs in the differential diagnosis and treatment of Cushing's syndrome (editorial). J Clin Endocrinol Metab 78:17, 1994
- Krenning EP, Kwekkeboom DJ, Bakker WH, et al: Somatostatin receptor scintigraphy with [111 In-DTPA-D-Phe1 and [123 I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med 20:716, 1993
- Gold PW, Loriaux DL, Roy A, et al: Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing's disease. N Engl J Med 314:1329, 1986 [PMID 3010108]
- Groote Veldman R, Meinders AE: On the mechanism of alcohol-induced pseudo-Cushing's syndrome. Endocr Rev 17:262, 1996 [PMID 8771359]
- Shapiro MS, Shenkman L: Variable hormonogenesis in Cushing's syndrome. Q J Med 288:351, 1991
- Guilhaume B, Bertagna X, Thomsen M, et al: Transsphenoidal pituitary surgery for the treatment of Cushing's disease: results in 64 patients and long term follow-up studies. J Clin Endocrinol Metab 66:1056, 1988 [PMID 3360898]
- Melby JC: Therapy of Cushing disease: a consensus for pituitary microsurgery. Ann Intern Med 109:445, 1988
- Mampalam TJ, Tyrrell JB, Wilson CB: Transsphenoidal microsurgery for Cushing disease. Ann Intern Med 109:487, 1988 [PMID 2843068]
- Chandler WF, Schteingart DE, Lloyd RV, et al: Surgical treatment of Cushing's disease. J Neurosurg 66:204, 1987 [PMID 3806203]
- Lamberts SWJ, van der Lely AJ, de Herder WW: Transsphenoidal selective adenomectomy is the treatment of choice in patients with Cushing's disease: considerations concerning preoperative medical treatment and the long-term follow-up (editorial). J Clin Endocrinol Metab 80:3111, 1995
- Tahir AH, Sheeler LR: Recurrent Cushing's disease after transsphenoidal surgery. Arch Intern Med 152:977, 1992 [PMID 1580725]
- Avgerinos PC, Nieman LK, Oldfield EH, et al: The effect of pulsatile human corticotropin-releasing hormone administration on the adrenal insufficiency that follows cure of Cushing's disease. J Clin Endocrinol Metab 68:912, 1989 [PMID 2541160]
- Sarkar R, Thompson NW, McLeod MK: The role of adrenalectomy in Cushing's syndrome. Surgery 108:1079, 1990 [PMID 2247833]
- Doherty GM, Nieman LK, Cutler GB Jr, et al: Time to recovery of the hypothalamic-pituitary-adrenal axis after curative resection of adrenal tumors in patients with Cushing's syndrome. Surgery 108:1085, 1990 [PMID 2247834]
- Grua JR, Nelson DH: ACTH-producing pituitary tumors. Endocrinol Metab Clin North Am 20:319, 1991
- Axelrod L: Glucocorticoid therapy. Medicine (Baltimore) 55:39, 1976
-
Salem M, Tainsh RE Jr, Bromberg J, et al: Perioperative
glucocorticoid coverage: a reassessment 42 years after emergence of a problem.Ann
Surg 219:416, 1994 [PMID
8161268]
-
May ME, Carey RM: Rapid adrenocorticotropic hormone test
in practice. Am J Med 79:679, 1985
-
Streeten DHP, Anderson GH Jr, Bonaventura MM: The potential
for serious consequences from misinterpreting normal responses to the rapid
adrenocorticotropin test. J Clin Endocrinol Metab 81:285, 1996
-
Gittler RD, Fajans SS: Primary aldosteronism (Conn's
syndrome). J Clin Endocrinol Metab 80:3438, 1995
-
White PC: Disorders of aldosterone biosynthesis and
action. N Engl J Med 331:250, 1994
-
Blumenfeld JD, Sealey JE, Schlussel Y, et al: Diagnosis
and treatment of primary hyperaldosteronism. Ann Intern Med 121:877, 1994
[PMID
7978702]
-
Bravo EL: Primary aldosteronism: issues of diagnosis
and management. Endocrinol Metab Clin North Am 23:271, 1994
-
Litchfield WR, Dluhy RG: Primary aldosteronism. Endocrinol
Metab Clin North Am 24:593, 1995
-
Melby JC: Diagnosis of hyperaldosteronism. Endocrinol
Metab Clin North Am 20:247, 1991
-
. McKenna TJ, Sequeira SJ, Heffernan A, et al: Diagnosis
under random conditions of all disorders of the renin-angiotensin-aldosterone
axis, including primary hyperaldosteronism. J Clin Endocrinol Metab 73:952,
1991 [PMID
1939533]
-
Dluhy RG, Lifton RP: Glucocorticoid-remediable aldosteronism.
Endocrinol Metab Clin North Am 23:285, 1994
-
McCroskey RD, Phillips A, Mott F, et al: Antiphospholipid
antibodies and adrenal hemorrhage. Am J Hematol 36:60, 1991 [PMID
1984684]
-
Asherson RA, Hughes GR: Hypoadrenalism, Addison's disease
and antiphospholipid antibodies (editorial). J Rheumatol 18:1, 1991 [PMID
2023176]
-
Claussen MS, Landercasper J, Cogbill TH: Acute adrenal
insufficiency presenting as shock after trauma and surgery: three cases
and review of the literature. J Trauma 32:94, 1992 [PMID
1732582]
-
Rao RH, Vagnucci AH, Amico JA: Bilateral massive adrenal
hemorrhage: early recognition and treatment. Ann Intern Med 110:227, 1989
[PMID
2643380]
-
Fitzpatrick PM, Swensen SJ: Report of an unusual case
of postoperative adrenal hemorrhage in a young man. Am J Med 86:487, 1989
[PMID
2929638]
-
Szalados JE, Vukmir RB: Acute adrenal insufficiency
resulting from adrenal hemorrhage as indicated by post-operative hypotension.
Intensive Care Med 20:216, 1994 [PMID
8014290]
-
Grinspoon SK, Bilezikian JP: HIV disease and the endocrine
system. N Engl J Med 327:1360, 1992 [PMID
1406838]
-
Leinung MC, Liporace R, Miller CH: Induction of adrenal
suppression by megestrol acetate in patients with AIDS. Ann Intern Med
122:843, 1995 [PMID
7741369]
-
Loprinzi CL: Effect of megestrol acetate on the human
pituitary-adrenal axis. Mayo Clin Proc 67:1160, 1992
-
Oster MH, Enders SR, Samuels SJ, et al: Megestrol acetate
in patients with AIDS and cachexia. Ann Intern Med 121:400, 1994 [PMID
8053613]
-
Vita JA, Silverberg SJ, Goland RS, et al: Clinical
clues to the cause of Addison's disease. Am J Med 78:461, 1985 [PMID
3976705]
-
Kyriazopoulou V, Parparousi O, Vagenakis AG: Rifampicin-induced
adrenal crisis in addisonian patients receiving corticosteroid replacement
therapy. J Clin Endocrinol Metab 59:1204, 1984 [PMID
6490796]
-
Wagner RL, White PF, Kan PB, et al: Inhibition of adrenal
steroidogenesis by the anesthetic etomidate. N Engl J Med 310:1415, 1984
[PMID
6325910]
-
Tucker WS Jr, Snell BB, Island DP, et al: Reversible
adrenal insufficiency induced by ketoconazole. JAMA 253:2413, 1985 [PMID
3981770]
-
Seidenwurm DJ, Elmer EB, Kaplan LM, et al: Metastases
to the adrenal glands and the development of Addison's disease. Cancer
54:552, 1984 [PMID
6733685]
-
Grinspoon SK, Biller BMK: Clinical Review 62: laboratory
assessment of adrenal insufficiency. J Clin Endocrinol Metab 79:923, 1994
[PMID
7962298]
-
Fiad TM, Kirby JM, Cunningham SK, et al: The overnight
single-dose metyrapone test is a simple and reliable index of the hypothalamic-pituitary-adrenal
axis. Clin Endocrinol 40:603, 1994
-
Hermus ARMM, Pieters GFFM, Pesman GJ, et al: CRH as
a diagnostic and heuristic tool in hypothalamic-pituitary diseases. Horm
Metab Res (suppl) 16:68, 1987
-
Ulick S, Wang JZ, Morton DH: The biochemical phenotypes
of two inborn errors in the biosynthesis of aldosterone. J Clin Endocrinol
Metab 72:1415, 1992
-
Jagger PI: Hypoaldosteronism. Endocrinologist 5:23,
1995
-
Oster JR, Singer I, Fishman LM: Heparin-induced aldosterone
suppression and hyperkalemia. Am J Med 98:575, 1995 [PMID
7778574]
-
Leshin M: Southwestern Internal Medicine Conference:
polyglandular autoimmune syndromes. Am J Med Sci 290:77, 1985
-
Trence DL, Morley JE, Handwerger BS: Polyglandular
autoimmune syndromes. Am J Med 77:107, 1984
-
Loriaux DL: The polyendocrine deficiency syndromes
(editorial). N Engl J Med 312:1568, 1985
-
Ahonen P, Myllarniemi S, Sipila I, et al: Clinical
variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
(APECED) in a series of 68 patients. N Engl J Med 322:1829, 1990 [PMID
2348835]
-
Werk EE Jr, Sholiton LJ, Kalejs L: Testosterone-secreting
adrenal adenoma under gonadotropin control. N Engl J Med 289:767, 1973
[PMID
4728749]
-
Nakano M: Adrenal cortical carcinoma. Acta Pathol Jpn
38:163, 1988
-
Givens JR, Andersen RN, Wiser WL, et al: A gonadotropin-responsive
adrenocortical adenoma. J Clin Endocrinol Metab 38:126, 1974 [PMID
4358642]
-
Larson BA, Vanderlaan WP, Judd HL, et al: A testosterone-producing
adrenal cortical adenoma in an elderly woman. J Clin Endocrinol Metab 42:882,
1976 [PMID
1270580]
-
Schteingart DE, Woodbury MC, Tsao HS, et al: Virilizing
syndrome associated with an adrenal cortical adenoma secreting predominantly
testosterone. Am J Med 67:140, 1979 [PMID
223440]
-
Gabrilove JL, Sharma DC, Wotiz HH, et al: Feminizing
adrenocortical tumors in the male: a review of 52 cases including a case
report. Medicine (Baltimore) 44:37, 1965
-
Desai MB, Kapadia SN: Feminizing adrenocortical tumors
in male patients: adenoma versus carcinoma. J Urol 139:101, 1988 [PMID
3336072]
-
Reincke M, Nieke J, Krestin GP, et al: Preclinical
Cushing's syndrome in adrenal "incidentalomas": comparison with adrenal
Cushing's syndrome. J Clin Endocrinol Metab 75:826, 1992
-
Herrera MF, Grant CS, van Heerden JA, et al: Incidentally
discovered adrenal tumors: an institutional perspective. Surgery 110:1014,
1991 [PMID
1745970]
-
Rosen HN, Swartz SL: Subtle glucocorticoid excess in
patients with adrenal incidentaloma. Am J Med 92:213, 1992 [PMID
1543208]
-
Bernini G, Sgro M, Molea N, et al: A documented clinical
case of pre-Cushing's syndrome. Endocrinologist 5:377, 1995
-
Osella G, Terzolo M, Borretta G, et al: Endocrine evaluation
of incidentally discovered adrenal masses (incidentalomas). J Clin Endocrinol
Metab 79:1532, 1994
-
Cook DM, Loriaux DL: The incidental adrenal mass. Endocrinologist
6:4, 1996
-
Staren ED, Prinz RA: Selection of patients with adrenal
incidentalomas for operation. Surg Clin North Am 75:499, 1995 [PMID
7747255]
-
White PC, New MI: Genetic basis of endocrine disease
2: congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin
Endocrinol Metab 74:6, 1992 [PMID
1727830]
-
Azziz R, Dewailly D, Owerbach D: Clinical review 56:
Nonclassic adre nal hyperplasia: current concepts. J Clin Endocrinol Metab
78:810, 1994 [PMID
8157702]
-
Rittmaster RS: Hyperandrogenism-what is normal? (editorial).
N Engl J Med 327:194, 1992
-
Fruzzetti F, De Lorenzo D, Parrini D, et al: Effects
of finasteride, a 5a-reductase inhibitor, on
circulating androgens and gonadotropin secretion in hirsute women. J Clin
Endocrinol Metab 79:831, 1994 [PMID
8077369]
-
Moghetti P, Castello R, Magnani CM, et al: Clinical
and hormonal effects of the 5a-reductase inhibitor
finasteride in idiopathic hirsutism. J Clin Endocrinol Metab 79:1115, 1994
-
Derksen J, Nagesser SK, Meinders AE, et al: Identification
of virilizing adrenal tumors in hirsute women. N Engl J Med 331:968, 1994
[PMID
8084355]
-
McKenna TJ: Screening for sinister causes of hirsutism
(editorial). N Engl J Med 331:1015, 1994
-
Wortsman J, Frank S, Cryer PE: Adrenomedullary response
to maximal stress in humans. Am J Med 77:779, 1984 [PMID
6496531]
-
The function of adrenaline (editorial). Lancet 1:561,
1985
-
Daly PA, Landsberg L: Phaeochromocytoma: diagnosis
and management. Bailliere's Clin Endocrinol Metab 6:143, 1992
-
Golub MS, Tuck ML: Diagnostic and therapeutic strategies
in pheochromocytoma. Endocrinologist 2:101, 1992
-
Whalen RK, Althausen AF, Daniels GH: Extra-adrenal
pheochromocytoma. J Urol 147:1, 1992
- Havlik RJ, Cahow CE, Kinder BE: Advances in the diagnosis and treatment of pheochromocytoma. Arch Surg 123:626, 1988 [PMID 3282495]