Contenido del artículo
X INSUFICIENCIA RENAL CRONICA
- Progresión de la enfermedad renal: Patogenia y prevención
- HIPERFILTRACION, HIPERTENSION GLOMERULAR E HIPERTENSION SISTEMICA
- FACTORES DIETETICOS
- CONTROL DE LA HIPERGLUCEMIA
- FACTORES DE CRECIMIENTO
- Adaptación a la insuficiencia renal crónica
- Estudio del paciente con insuficiencia renal crónica
DRA. NINA E. TOLKOFF-RUBIN
DR. MANUEL PASCUAL
La insuficiencia renal crónica es un estado de deterioro progresivo e irreversible de la función renal como consecuencia de un amplio espectro de enfermedades [ver tabla 1]. Aunque con frecuencia existen evidencias del proceso patológico primario, la etiología es difícil de determinar en la insuficiencia renal avanzada. Las manifestaciones clínicas de la insuficiencia renal crónica son independientes del proceso patológico inicial que afectó a los riñones y en cambio reflejan la incapacidad general de los riñones para excretar los desechos nitrogenados, regular el equilibrio hidroeléctrico y secretar hormonas.
|
|
|
Progresión de la enfermedad renal: Patogenia y prevención
En la mayoría de los pacientes con enfermedad renal crónica, independientemente de su etiología, la reducción de la velocidad de filtración glomerular (VFG) a menos de 25 ml/min se caracteriza por deterioro progresivo de la función renal que lleva eventualmente a insuficiencia renal terminal con necesidad de diálisis o trasplante. Esta pérdida de la función renal suele ocurrir a una velocidad constante en cada paciente y continúa incluso cuando el proceso que inició el daño renal (como un proceso inmunológico, uropatía obstructiva, diabetes, hipertensión o nefropatía tóxica) no está ya activo.1 Por lo tanto, parece ser que después de la pérdida inicial de la función renal se desencadenan nuevos mecanismos fisiopatogénicos no relacionados con la enfermedad primaria, que son responsables de la progresión inexorable a la insuficiencia renal terminal.1-6
HIPERFILTRACION, HIPERTENSION GLOMERULAR E HIPERTENSION SISTEMICA
Varios estudios experimentales han incriminado las adaptaciones hemodinámicas intrarrenales como principales culpables en la progresión de la enfermedad renal.4 Se ha supuesto que en respuesta a un daño importante en el riñón, las nefronas restantes sufren cambios estructurales y funcionales que promueven la lesión glomerular, intersticial o ambas. Pronto después de que ocurre el daño inicial al riñón, la hiperfunción e hipertrofia de las nefronas restantes parecen ser benéficas porque tienden a normalizar la función renal. Sin embargo, en los modelos de ablación renal en ratas estas adaptaciones tienen efectos dañinos a largo plazo porque puede presentarse glomeruloesclerosis focal y segmentaria y enfermedad renal terminal.1,2
¿Cómo puede la ablación renal provocar estos cambios? Aunque no se ha definido el mecanismo preciso, un modelo animal ha demostrado que la nefrectomía parcial causa reducción en la resistencia vascular preglomerular en las nefronas restantes.2 Esta disminución en la resistencia vascular preglomerular provoca aumento en el flujo plasmático en los capilares glomerulares y un aumento concomitante en el gradiente de presión a través de las paredes capilares glomerulares (hipertensión glomerular). El resultado neto de estos cambios es que ocurre hiperfiltración en las nefronas restantes, lo que origina cicatrización progresiva del glomérulo.1,2 La hipertensión sistémica, la diabetes mellitus y otras enfermedades renales pueden asociarse con adaptaciones hemodinámicas intrarrenales que son semejantes a las observadas en animales parcialmente nefrectomizados [ver figura 1].
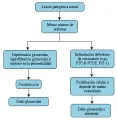
|
| Figura 1 |
| Patogenia de la enfermedad renal progresiva |
También se ha identificado a la hipertensión sistémica como un factor de riesgo importante en la progresión de la enfermedad renal.7,8 Su papel es especialmente importante por la alta incidencia de hipertensión en pacientes que sufren de insuficiencia renal crónica de diversas causas. Cuando la hipertensión sistémica no se controla (i.e., existe hipertensión acelerada o maligna), puede ocurrir pérdida rápida y progresiva de la función renal. Por lo tanto, es muy importante la vigilancia estrecha de la presión arterial en pacientes con insuficiencia renal crónica.
La hipertensión sistémica también puede ser la causa principal de la insuficiencia renal, en especial en afroamericanos.8,9 Los factores genéticos pueden ser críticos para determinar si se desarrolla enfermedad renal en un paciente hipertenso.9 Esta hipótesis es apoyada por el hecho de que la hipertensión leve a moderada de larga evolución rara vez se asocia con pérdida progresiva de la función renal.8-10
Puede lograrse reducción de la hipertensión glomerular, así como de la hipertensión sistémica, administrando inhibidores de la enzima convertidora de angiotensina (ECA).8,11 Esta reducción, que causa vasodilatación de la arteriola eferente, es mediada por menor formación de angiotensina II, que regula el tono posglomerular de la arteriola eferente.11 Sin embargo, estas modificaciones de la hemodinámica intraglomerular proporcionan solo una explicación parcial para el efecto renoprotector de los inhibidores de la ECA.
Los estudios realizados en ratas parcialmente nefrectomizadas han demostrado que la administración de un inhibidor de la ECA también reduce de modo significativo la proteinura y la progresión de lesiones escleróticas.8,11 Por el contrario, un esquema triple de otros antihipertensivos que disminuya la presión arterial sistémica a un nivel similar al logrado con la inhibición de la ECA no afecta la excreción proteica o la progresión de la lesión renal. Estudios recientes en humanos confirmaron estos efectos benéficos. En 1993, los resultados de un estudio de cuatro años dirigido por el Collaborative Study Group de los EUA demostró que el inhibidor de la ECA captopril disminuye en forma significativa la velocidad de progresión de la insuficiencia renal en pacientes con diabetes mellitus insulino dependiente (DMID).12 Aún más, el número de pacientes que fallecieron o requirieron diálisis o trasplante fue 50 porciento menor en el grupo tratado con captopril que el del grupo tratado con placebo. Los resultados de este estudio prospectivo, aleatorio y controlado con placebo indican en forma clara que el captopril puede proteger la función renal en pacientes con DMID. Además, sugieren que el efecto renoprotector del captopril no solo se debe al control de la presión arterial sistémica, porque ambos grupos del estudio tuvieron presiones semejantes.12 En los pacientes no diabéticos, los inhibidores de la ECA fueron superiores a los beta bloqueadores para retrasar la progresión de la insuficiencia renal.13
Por lo tanto, los datos de estudios clínicos y experimentales indican que todos los antihipertensivos no tienen los mismos efectos y que propiedades únicas de los inhibidores de la ECA pueden ser las responsables de su efecto para retrasar la progresión del daño renal. La eficacia de los inhibidores de la ECA puede deberse no solo a sus propiedades hemodinámicas (i.e., su capacidad para disminuir la hipertensión tanto sistémica como glomerular) sino también a su capacidad para reducir la proteinuria y para interferir con la regulación de la expresión de factores de crecimiento críticos, como el factor de transformación de crecimiento-b (FTC-b)1,5,6 (ver adelante).
Los datos experimentales sugieren que los bloqueadores de los canales del calcio pueden también ser eficaces en caso de insuficiencia renal crónica.14,15 Estos medicamentos pueden disminuir la proteinuria y la lesión glomerular renal al inhibir el crecimiento renal compensatorio, disminuyendo la presión intraglomerular.14,15
FACTORES DIETETICOS
Durante muchos años los investigadores han sospechado que los factores dietéticos, en especial la ingesta de proteínas, tiene influencia en la progresión de la enfermedad renal.16,17 Los resultados de estudios que usan modelos animales han demostrado que la ingesta de proteínas puede causar hipertensión glomerular que puede ser dañina para el riñón.2,16,17 Después de la ingestión proteica, la presión capilar glomerular y la VFG típicamente aumentan, creando un estado de hiperfiltración.
En modelos animales de enfermedad renal, las dietas bajas en proteínas retrasan la progresión de la insuficiencia renal.2 Se ha sugerido que los efectos benéficos de la restricción proteica son causados por mejoría en la hemodinámica intraglomerular en las nefronas no lesionadas, lo que disminuye la proteinuria y retrasa la glomeruloesclerosis progresiva. Además, el contenido proteico de la dieta puede influir en la expresión de factores de crecimiento, como el FTC-b, que se relacionan con la cicatrización progresiva del riñón.18
Aunque existe evidencia intensa de que la restricción proteica retrasa la progresión de la insuficiencia renal en modelos experimentales, los datos de estudios en humanos son menos concluyentes. Varios estudios en pacientes diabéticos han sugerido que la restricción proteica tiene un efecto benéfico,19-23 pero hasta hace poco existían solo algunos estudios aleatorios y prospectivos que habían evaluado grandes poblaciones de pacientes no diabéticos y que usaron métodos sensibles para medir el cumplimiento y la VFG.
En 1994 se reportaron los muy esperados resultados del Estudio de Modificación de la Dieta en la Enfermedad Renal (MDRD), patrocinado por los Institutos Nacionales de Salud de los EUA.24 En este estudio se distribuyó en forma aleatoria a pacientes no diábeticos con insuficiencia renal moderada (VFG de 25 a 55 ml/min/1.73 m2) para recibir una dieta baja en proteínas (0.6 g/kg/día) o una dieta normal en proteínas (1.3 g/kg/día). Los pacientes con insuficiencia renal severa (VFG de 13 a 24 ml/min/1.73 m2) recibieron una dieta baja en proteínas (0.6 g/kg/día) o una dieta muy baja en proteínas (0.3 g/kg/día), ningún paciente en esta categoría recibió dieta normal.
A los tres años de seguimiento, este estudio prospectivo y multicéntrico no pudo demostrar con claridad que la restricción proteica beneficiara a los pacientes no diabéticos con insuficiencia renal moderada de diversas etiologías.24 Aunque los pacientes que recibieron la dieta restringida en proteínas tuvieron una reducción inicial rápida de la VFG (hallazgo semejante al observado en estudios en animales), la reducción posterior en la VFG fue más lenta en los pacientes con restricción proteica que en los que recibieron una dieta normal [ver figura 2]. Este dato sugiere que un seguimiento más prolongado puede revelar un mayor beneficio de la restricción proteica. También serán interesantes los resultados del análisis de los diferentes subgrupos.
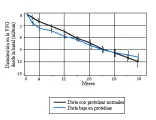
|
| Figura 2 |
| Efecto de las proteínas de la dieta en la VFG |
En los pacientes con insuficiencia renal severa que participaron en este estudio, la dieta muy baja en proteínas no se asoció con menor progresión del daño renal comparado con la dieta baja en proteínas.24 Sin embargo, el estudio MDRD sí muestra que disminuir la presión arterial (con cifra ideal de 125/75) redujo la progresión de la enfermedad renal en pacientes con proteinuria mayor de 1 g/día. La disminución de la presión arterial también produjo una tendencia hacia un mayor beneficio en los afroamericanos con insuficiencia renal moderada.
Se ha sugerido que otros factores dietéticos, en especial la ingesta de fosfato, también puede participar en la progresión del daño renal. Los resultados de estudios en animales han demostrado que la restricción de fosfatos en la dieta puede retrasar la progresión de la enfermedad renal, quizá por limitar la calcificación del tejido renal (nefrocalcinosis) que depende de la alta concentración de fosfato de calcio.25,26 Sin embargo, aún no existen evidencias definitivas en humanos de que la restricción en la ingesta de fosfato retrase la progresión de la nefropatía. Del mismo modo, muchos de los datos sobre el papel de los lípidos provienen de estudios en animales. Por ejemplo, un modelo de rata nefrectomizada ha demostrado que la intervención farmacológica para reducir la concentración de lípidos retrasa la progresión de la insuficiencia renal.16 Sin embargo, no existen evidencias clínicas controladas en humanos de que la restricción de lípidos en la dieta sea benéfica para la insuficiencia renal,16 y se requieren estudios clínicos prospectivos y aleatorios sobre dietas bajas en colesterol, tratamiento reductor de lípidos o ambos. Sin embargo, debido a la alta prevalencia de enfermedad cardiovascular en pacientes con enfermedad renal progresiva, parece prudente tratar la hiperlipidemia significativa en los pacientes que tengan un perfil de lípidos aterogénico.
CONTROL DE LA HIPERGLUCEMIA
Los resultados recientes del Estudio de Control y Complicaciones de la Diabetes (DCCT) de los EUA han demostrado en definitiva que el control intensivo de la glucemia en pacientes con DMID (i.e., con intento de mantener el nivel de hemoglobina A1c cerca del rango normal) puede retrasar de modo eficaz el inicio de la proteinuria y, más importante, la progresión de la nefropatía, retinopatía y neuropatía.27 El tratamiento intensivo requiere con frecuencia de la administración de tres o más inyecciones de insulina por día o del uso de una bomba de insulina, con vigilancia estrecha de la glucemia. Los resultados de este estudio apoyan la hipótesis de que la hiperglucemia está implicada en la patogenia de las complicaciones a largo plazo de la DMID y que el control estricto de la glucemia tiene un impacto importante para prevenir o retrasar el inicio de las complicaciones diabéticas.
FACTORES DE CRECIMIENTO
Durante los años recientes se ha vuelto evidente que los factores de crecimento, en especial el FTC-b, pueden tener importancia crucial en la fisiopatología de la enfermedad renal y pueden contribuir a la progresión de la insuficiencia renal.1,3,5,8,28 Por ejemplo, los estudios que emplearon un modelo de rata de glomerulonefritis inmune mostraron que el FTC-b es un mediador importante de la lesión renal.3-5,28 En este modelo ocurre proliferación de la matriz extracelular en el glomérulo, y la expresión de FTC-ß aumenta en forma simultánea. El papel critico del FTC-b en este modelo se ha demostrado por el hallazgo de que al administrar anticuerpos específicos contra el FTC-b puede prevenirse el desarrollo de enfermedad renal.3,5,28 Se ha observado expresión progresiva y constante del FTC-b en un modelo de glomerulonefritis crónica, que causa glomeruloesclerosis y proteinuria.3,5,28 Además, se ha implicado a la mayor expresión de FTC-b en la fibrosis intersticial que ocurre en la uropatía obstructiva,6,29 la nefropatía diabética,28 y en la glomerulonefritis focal asociada con infección por el virus de inmunodeficiencia humana (VIH).30
¿Cómo puede la expresión exagerada de FTC-b causar glomeruloesclerosis y fibrosis intersticial? El FTC-b es un polipéptido que tiene muchas acciones biológicas, incluyendo quimiotaxis, regulación de la síntesis de otros factores de crecimiento, inhibición de la función de células T y B, inducción e inhibición de la proliferación celular y fibrogénesis.5,6,28 La inducción de la formación y depósito de matriz extracelular es una característica constante de las varias acciones del FTC-b5,28: este factor de crecimiento estimula a las células para aumentar la síntesis de componentes de la matriz extracelular y puede disminuir la producción de proteasas degradadoras de la matriz, causando un desplazamiento del balance hacia la fibrogénesis.3,6,28,30 Además, recientemente se ha demostrado que la angiotensina II puede actuar como un factor de crecimiento que induce la expresión de FTC-b en células del músculo liso vascular y células mesangiales, causando hipertrofia de estas células.4,6,28,31 Por lo tanto, puede ser que la eficacia de los inhibidores de la ECA para prevenir la progresión de la insuficiencia renal derive no solo de los efectos de estos agentes sobre la hemodinamia sistémica e intraglomerular, sino también de regular en forma inhibitoria el FTC-b al reducir la angiotensina II.5,28,31 Sin duda las investigaciones actuales aclararán estas interesantes hipótesis.
Es posible que otras citocinas y factores de crecimiento jueguen un papel en la progresión inexorable de la enfermedad renal. Se ha identificado al factor de crecimiento derivado de plaquetas (FCDP), otro polipéptido multifuncional, como un mediador importante.1,32 Este es una glucoproteína que tiene potente actividad mitogénica y vasocactiva, y que regula la respuesta inflamatoria y el metabolismo extracelular,31 expresándose en varios tipos diferentes de células en el riñón.33 Un estudio reciente encontró que tanto el FCDP como el FTC-b median la producción de colágena por células mesangiales que son expuestas a niveles altos de glucosa o a productos terminales de la glucosilación. Este dato puede ser especialmente importante en la patogenia de la nefropatía diabética, que se caracteriza por depósito anormal de la matriz en el mesangio, causando glomeruloesclerosis progresiva.33 Otro factor de crecimiento peptídico, el factor de crecimiento semejante a insulina-1 (FCI-1), puede también contribuir a la glomeruloesclerosis. In vitro, el FCI-1 aumenta la producción de matriz mesangial y estimula la hiperplasia.34
Sin embargo, la inducción de un estado profibrogénico incluye no solo el depósito de matriz extracelular, sino también la desrregulación de las proteasas degradadoras. Estas enzimas son capaces de degradar tanto los componentes colágena como no colágena de la matriz extracelular y están íntimamente involucrados en la regulación de la remodelación tisular. Se ha sugerido que la disminución en estas enzimas o un aumento en los inhibidores de proteasa específicos de tejido pueden contribuir también a aumentar la fibrosis y la progresión de la enfermedad renal.6,29
Adaptación a la insuficiencia renal crónica
El riñón normal controla con precisión la concentración de solutos y el volumen de agua, a pesar de las grandes variaciones en la ingestión diaria. En las enfermedades renales crónicas los pacientes aún pueden excretar solutos y volúmenes de agua sin alterar significativamente su dieta, incluso cuando la velocidad de filtración glomerular (VFG) está muy disminuida. De hecho, muchos pacientes permanecen relativamente asintomáticos hasta que la función renal disminuye por debajo del 10 porciento de lo normal,35 lo que demuestra la capacidad de adaptación del riñón a la lesión. Para mantener la homeostasis del agua y los electrolitos cuando la ingestión no es regulada y hay una disminución del número de nefronas, la tasa de excreción de las nefronas aumentará mientras que el funcionamiento de los glomérulos permanezca estrechamente integrado (balance glomerulotubular).35-37
Existen tres mecanismos principales de adaptación al deterioro progresivo de la función renal. El primer mecanismo se observa con sustancias como la urea y la creatinina, cuya excreción urinaria depende en gran medida de la VFG. De esta manera, cuando la VFG disminuye, los niveles de estas sustancias empiezan a aumentar. Sin embargo, los niveles plasmáticos no son directamente proporcionales al grado del deterioro de la VFG; mejor dicho, la relación entre los niveles plasmáticos y la VFG sigue una relación hiperbólica. En las etapas iniciales de la insuficiencia renal existen cambios mínimos en la concentración sérica de creatinina, a pesar de la disminución de la VFG en más del 50 porciento; esta estabilidad se debe principalmente a la secreción en el túbulo proximal. Sin embargo, más allá de este punto, cuando la reserva renal se agota, incluso mínimas reducciones de la VFG producen elevaciones importantes de las concentraciones séricas de urea y creatinina. Los síntomas urémicos tienen un patrón similar: a pesar de la reduccción importante en la función renal, existe un prolongado estado asintomático que es seguido por el aumento en el número y la gravedad de los síntomas relacionados con la elevación de las concentraciones séricas de urea y creatinina.35,36
El segundo mecanismo de adaptación al deterioro renal progresivo se observa con solutos como el potasio. La concentración sérica de potasio se mantiene dentro de los límites normales hasta que la reducción de la VFG se aproxima al 10 porciento de lo normal, momento en el que puede presentarse hipercalemia. Normalmente, el potasio es filtrado del suero y reabsorbido en los túbulos proximales y el asa de Henle. Por lo tanto, el potasio urinario deriva principalmente de la secreción de los túbulos colectores. Cuando algunas nefronas se han dañado, las nefronas intactas aumentan la secreción de potasio por medio del aumento del flujo sanguíneo y el aumento de la carga de sodio a los túbulos colectores.38 Además, debido a que la secreción de aldosterona aumenta en los pacientes con insuficiencia renal, hay una pérdida mayor de potasio a través del tubo digestivo.38 Este sistema de mayor excreción gastrointestinal funciona razonablemente bien cuando la ingestión de potasio en la dieta es normal. Sin embargo, los pacientes con insuficiencia renal crónica pueden llegar a tener hipercalemia cuando la vía se somete a un esfuerzo adicional por una carga endógena de potasio liberado en forma aguda (v.gr., secundaria a rabdomiólisis o hemólisis) o por una carga exógena (v.gr., por el aumento en el consumo de frutas y vegetales, la administración de sales de potasio o el uso de ciertos fármacos).
El tercer mecanismo de adaptación se observa en la homeostasis del sodio y la regulación del volumen extracelular. Comparado con los niveles de otros solutos, el balance de sodio permanece por lo general normal a lo largo de la evolución de la insuficiencia renal, a pesar de una ingestión constante,35 reflejando una impresionante adaptación biológica a la lesión. La tasa de la excreción del sodio depende de la cantidad de la carga filtrada que no es reabsorbida. Cuando la VFG disminuye, la filtración de la carga de sodio también lo hace. Sin embargo, la excreción fraccional de sodio aumenta notablemente para mantener la homeostasis. De hecho, la excreción fraccional de sodio en la insuficiencia renal crónica puede aumentar desde el uno al 25 porciento.35,36 Es evidente que este mecanismo es eficaz en la adaptación a la VFG disminuida sin modificar el consumo de sodio en la dieta; los pacientes pueden mantener el balance de sodio, incluso en etapas avanzadas de las enfermedades renales, si disminuyen gradualmente el consumo de sodio.39 No obstante, la capacidad de adaptación de los riñones a la insuficiencia renal progresiva es limitada. El sistema puede ser sobrepasado por una restricción sódica severa, lo que lleva a la depleción del volumen del líquido extracelular o por una saturación de sodio, que da lugar a sobrecarga del volumen.
Estudio del paciente con insuficiencia renal crónica
Una vez establecida la insuficiencia renal crónica, independientemente de su etiología, se presenta un inexorable deterioro de la función en las nefronas restantes. La depuración de inulina y de diversos compuestos radiactivos, como el iotalamato (que es más fácil de medir que la inulina en suero y en orina), parece ser el método más sensible disponible para medir la VFG y evaluar la progresión de la insuficiencia renal.40,41 Sin embargo, aunque estas técnicas deben ser el estándar de oro para evaluar los cambios en la función renal en los estudios clínicos, la medición de la creatinina sérica y de la depuración de creatinina proporcionan un índice útil de la VFG en la práctica. Estas pruebas siguen siendo una herramienta útil para el clínico por su comodidad y bajo costo, además de que pueden realizarse y repetirse con facilidad, lo que permite vigilar en forma estrecha los cambios significativos en la función renal.1,40-42 Un cambio repentino en la concentración de creatinina sérica debe hacer sospechar la posibilidad de un proceso agudo potencialmente reversible como uropatía obstructiva, factores prerrenales, hipercalcemia y respuestas adversas a los medicamentos (nefrotoxicidad o nefritis intersticial aguda), o enfermedad renovascular bilateral importante. Esta última debe sospecharse en cualquier paciente con hipertensión resistente al tratamiento e insuficiencia renal, debido a que la angioplastía y la revascularización quirúrgica pueden mejorar la función renal.43 Por esta razón debe considerarse la angiografía.
De este modo, el estudio inicial del paciente con insuficiencia renal crónica debe orientarse hacia la identificación de cualquier elemento potencialmente reversible que pueda contribuir a la insuficiencia renal o a precipitar su progresión44 [ver tabla 2]. Después es importante dirigir el tratamiento hacia las consecuencias del deterioro de la función renal.
|
|
ECA- enzima convertidora de angiotensina, VFG- velocidad de filtración glomerular *Deben vigilarse los niveles de potasio. Los inhibidores de la ECA están contraindicados en pacientes embarazadas y deben emplearse con precaución en enfermos con lesión bilateral de la arteria renal y en los que tienen un solo riñón. |
SINDROME UREMICO
Patogenia
El síndrome urémico es un conjunto de signos y síntomas producidos por la incapacidad progresiva del riñón para realizar las funciones de excreción, secreción y regulación. Estos síntomas incluyen anorexia, náusea, vómito, laxitud, prurito, espasmos mioclónicos (temblor involuntario muscular), anemia y tendencia hemorragípara. La búsqueda de las llamadas toxinas urémicas continúa en un intento por explicar las múltiples manifestaciones que caracterizan al síndrome urémico y que afecta virtualmente a todo el organismo. Se ha implicado a muchas sustancias (v.gr., fenoles, ácidos orgánicos, urea, guanidina y las llamadas moléculas medianas, que tienen un peso molecular de cerca de 500 a 2,000 daltons), pero no se ha identificado un compuesto único que reproduzca el cuadro clínico completo.45 Aunque todavía se duda del hecho de que la urea por sí misma produzca las alteraciones observadas (excepto en concentraciones muy altas), los niveles de nitrógeno ureico en sangre (BUN) constituyen un indicador clínico útil de la gravedad del estado urémico y de la respuesta de los pacientes al tratamiento.45 Por el contrario, los niveles séricos de creatinina, aunque constituyen un buen indicador de la VFG, correlacionan poco con los síntomas urémicos.
Tratamiento
Durante más de un siglo el tratamiento de insuficiencia renal crónica y de los síntomas urémicos se ha centrado en la restricción proteica en la dieta. Debido a que la mayoría de las posibles toxinas urémicas se derivan del metabolismo proteico, se considera que al disminuir el consumo de proteínas se disminuirá la formación de estas sustancias,46 y por lo tanto los síntomas urémicos mejorarán. Algunos investigadores han demostrado que una dieta baja en proteínas (0.6 g/kg/día o 20 a 25 g de proteínas de alto valor biológico con suplementos de aminoácidos esenciales) produce disminución de los niveles de BUN, disminuyendo la anorexia, náusea, vómito, y convierte el balance nitrogenado negativo en positivo.47-49 La eficacia de una dieta baja en proteínas se debe a la reducción del catabolismo proteico y de la velocidad de formación de urea.
HIPERCALEMIA
Patogenia y diagnóstico
En los pacientes con insuficiencia renal aguda oligúrica, la hipercalemia es una urgencia médica que normalmente requiere tratamiento inmediato para evitar las complicaciones que ponen en peligro la vida, como el paro cardiaco y la parálisis muscular. Por el contrario, los riñones de los pacientes con insuficiencia renal crónica pueden adaptarse aumentando la secreción de potasio. En general, el desarrollo de la hipercalemia es más gradual y mejor tolerado en las etapas tardías de la evolución de la enfermedad, siempre y cuando se restringa el potasio en la dieta (generalmente a menos de 2 g/día) y se eviten los fármacos que afectan a la homeostasis del potasio. Hay tres tipos de fármacos que afectan la homeostasis del potasio: 1) fármacos que proporcionan un aporte exógeno de potasio, como los complementos de sales de potasio y la penicilina potásica, 2) fármacos que afectan la excreción del potasio, como el triamterene, la espironolactona y los antiinflamatorios no esteroideos, y 3) fármacos que inhiben el sistema renina-angiotensina-aldosterona, como los beta bloqueadores y los inhibidores de la ECA.38
Cualquier paciente con insuficiencia renal crónica cuyo consumo dietético de potasio, especialmente en frutas y verduras, exceda la tasa de excreción, puede evolucionar peligrosamente hacia la hipercalemia. Además, la acidosis severa, las infecciones agudas con una respuesta catabólica importante, la rabdomiolisis, la hemólisis aguda, la hiperglucemia severa o cualquier complicación añadida que causen oliguria pueden producir el desarrollo rápido de hipercalemia que pone en peligro la vida de los pacientes con insuficiencia renal crónica.
La hipercalemia también se presenta en los pacientes con insuficiencia renal leve o moderada, en las nefritis intersticiales o en los pacientes con diabetes mellitus.38 En muchos de estos pacientes la secreción de renina y aldosterona están disminuidas y no responden al estímulo del volumen. En algunos pacientes el defecto primario es el deterioro en la liberación de renina. Otros pacientes tienen, al parecer, un defecto a nivel de las suprarrenales que inhibe la síntesis de aldosterona.38 El resultado es la disminución de la reabsorción de sodio y de la secreción de potasio en los túbulos colectores, produciendo de esta manera la hipercalemia. Además, los pacientes con alteraciones en las glándulas suprarrenales pueden presentar una respuesta tubular inadecuada a los mineralocorticoides. Pueden requerirse dosis elevadas de fludrocortisona (0.4 a 1.0 mg/día) para corregir el defecto (la dosis normal de sustitución es de 0.1 mg/día).38
|
||||||||||||||||||||||||||||
IZC-insulina zinc cristalina
|
Los cambios electrocardiográficos (ECG) continúan siendo el mejor método de vigilancia para instituir el tratamiento. La primera alteración que se observa es la presencia de ondas T acuminadas; si esta alteración no se trata, los cambios ECG progresan y se manifiestan por ensanchamiento del complejo QRS y del intervalo P-R, con aplanamiento de las ondas P. Se puede presentar paro cardiaco con fibrilación ventricular o bloqueo.
Tratamiento
Si los niveles de potasio son superiores a 6.5 mEq/L o se presenta cualquiera de los cambios ECG mencionados, se debe iniciar de inmediato el tratamiento de la hipercalemia [ver tabla 2]. El tratamiento inicial de la hipercalemia debe dirigirse a antagonizar los efectos del potasio en el corazón (mediante la administración intravenosa de calcio) y a facilitar el transporte del potasio al interior de la célula en forma rápida (mediante la infusión de una solución de glucosa, insulina y bicarbonato).
Debido a que estas medidas para el control de la hipercalemia sólo son transitorias, simultáneamente deben comenzarse esfuerzos para eliminar el potasio del organismo. El potasio puede eliminarse eficazmente mediante la administración de iones de intercambio, como son las resinas de poliestireno de sulfonato sódico (25 a 50 g), junto con sorbitol por vía oral o como enema de retención para estimular un proceso diarreico. Aproximadamente se extrae 1 mEq de potasio por cada gramo de resina administrada, y por cada miliequivalente de potasio eliminado mediante el intercambio iónico de la resina se absorbe 1 mEq de sodio, por lo que se debe tener en cuenta la sobrecarga de sodio. Si estas medidas conservadoras no reducen adecuadamente los niveles séricos de potasio, se debe realizar diálisis (ver adelante).
ACIDOSIS
Patogenia
Los riñones eliminan 50 a 100 mEq/día de ácido producido por el metabolismo de las proteínas de la dieta. En la insuficiencia renal crónica las nefronas intactas mantienen esta excreción aumentando la producción de amonio hasta que la velocidad de filtración glomerular disminuye a menos de 40 a 50 ml/minuto.35,36 Por debajo de este punto el tejido renal indemne no puede compensarlo completamente y se presenta acidosis metabólica como resultado de la disminución de la excreción de amonio y fosfato y del acúmulo de ácidos orgánicos, lo que trae como consecuencia el desarrollo de acidosis metabólica con aumento de la brecha aniónica.
En contraste, algunos pacientes con insuficiencia renal leve o moderada secundaria a una enfermedad tubulointersticial crónica pueden desarrollar acidosis hiperclorémica sin aumento de la brecha aniónica; esta entidad parece ser resultado del deterioro en la capacidad de los túbulos renales para aumentar la producción de amonio en las etapas iniciales de la enfermedad.35
Tratamiento
La acidosis metabólica crónica (pH sérico < 7.3) produce síntomas como fatiga y letargo, aumento del esfuerzo respiratorio y sensibilidad a las catecolaminas, estimula la degradación de las proteínas50 y agrava la osteodistrofia renal.51 Por consiguiente, debe administrarse bicarbonato de sodio (1.2 a 2.4 g/día por vía oral) para mantener los niveles séricos de bicarbonato entre 18 y 20 mEq/L. Debido a que el citrato aumenta notablemente la absorción intestinal de aluminio, se debe evitar el uso de citrato sódico en los pacientes con insuficiencia renal crónica que toman fijadores de fosfato a base de aluminio.52
Se debe tener cuidado y evitar una corrección demasiado rápida de la acidosis, en particular si existe hipocalcemia. La acidosis disminuye la excitabilidad neuromuscular y protege contra los efectos de las concentraciones séricas demasiado bajas de calcio; de esta manera, los pacientes con insuficiencia renal pueden tener una hipocalcemia importante sin datos clínicos de tetania. Cuando la acidosis se corrige con demasiada rapidez, los niveles séricos de calcio ionizado disminuyen como resultado de la unión del calcio libre a las proteínas. Debido a que el calcio libre ionizado es muy importante para la función neuromuscular, la hipocalcemia puede desencadenar convulsiones.53
OSTEODISTROFIA RENAL
Patogenia y diagnóstico
La osteodistrofia renal es la complicación más importante de la insuficiencia renal crónica y es el resultado de una compleja interacción entre hiperparatiroidismo secundario, disminución de la producción renal de vitamina D y, en algunos pacientes, toxicidad por aluminio54-59 [ver figura 4]. A medida que disminuye la VFG, existe un descenso en la eliminación del fosfato y un aumento en la concentración sérica del mismo, que ocasiona disminución recíproca de la concentración del Ca2+ sérico. La hipocalcemia estimula la secreción de la hormona paratiroidea (PTH), produciendo resorción ósea (imagen histológica que corresponde a osteítis fibrosa) y liberación de calcio.
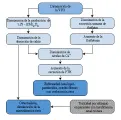
|
| Figura 4 |
| Patogenia de la osteodistrofia renal |
Un segundo factor que contribuye al metabolismo anormal del calcio y a la enfermedad ósea de tipo urémico es la disminución de la producción renal de 1,25-dihidroxivitamina D3 (1,25-(OH)2D3) o calcitriol, que es la forma activa de la vitamina D. El riñón con pérdida del parénquima funcional tiene una menor capacidad para convertir la 25-hidroxivitamina D3 [25-(OH)2D3] en el metabolito polar más activo 1,25-(OH)2D3. Esta hidroxilación también puede ser suprimida por los niveles tisulares elevados de fosfato. Como resultado de la disminución de la producción renal de 1,25-(OH)2D3, la absorción intestinal de calcio decrece, causando hipocalcemia, que disminuye la mineralización ósea y aumenta el estímulo para la liberación de PTH.54,56
La enfermedad ósea hiperparatiroidea es la forma más frecuente de osteodistrofia urémica; se diagnostica desde el punto de vista radiológico por la presencia de una notable desmineralización ósea con resorción de las clavículas y la imagen llamada en "sal y pimienta" del esqueleto, además de resorción subperióstica que se identifica con mayor facilidad en la porción radial de la falange media del dedo índice y de los dedos medios. El extremo de la falange distal también puede estar erosionado.54,55 En los pacientes con hiperparatiroidismo severo, cuando el producto de las concentraciones de fosfatos y los iones de calcio están muy elevados (por lo general más de 70), pueden presentarse calcificaciones metastásicas en las arterias y los tejidos blandos, que se manifiestan desde el punto de vista clínico por lesiones dérmicas dolorosas y necrosis isquémica de los dedos, o como depósitos periarticulares.
El diagnóstico de hiperparatiroidismo se confirma por los niveles séricos elevados de PTH. Sin embargo, es importante reconocer que la interpretación de los niveles de PTH en los pacientes con insuficiencia renal crónica depende del tipo de prueba usado para su determinación. Las pruebas para detectar la porción C-terminal de la PTH no son adecuadas, debido a que no sólo cuantifican la hormona activa, sino también los metabolitos inactivos. Puesto que estos metabolitos son excretados en condiciones normales por el riñón, los niveles de PTH cuantificados por este tipo de pruebas son, por lo general, mayores en los pacientes con insuficiencia renal crónica, independientemente del estado de las glándulas paratiroideas. Las pruebas para detectar la porción N-terminal de la PTH reflejan mejor la actividad de la paratiroides en los niveles esqueléticos, debido a que sólo se cuantifica la hormona activa.56 Aunque se pueden observar valores del doble de los normales en los pacientes bien compensados, los valores superiores a cinco o diez veces más sugieren la presencia de hiperparatiroidismo clínicamente importante.56
Otra forma de enfermedad ósea identificada en los pacientes con insuficiencia renal crónica, sobre todo en los pacientes sometidos a diálisis, es la secundaria al acúmulo de aluminio. Estos pacientes presentan con frecuencia dolor óseo grave e incapacitante, fracturas y debilidad.56 Aunque los niveles basales de aluminio, medidos por espectroscopía de absorción atómica, pueden proporcionar información útil si los niveles de aluminio están aumentados (>100 µg/ml), se considera a la prueba de la desferoxamina como el mejor indicador del depósito de aluminio corporal total.55,57 La desferroxamina es un compuesto quelante que libera aluminio de los depósitos corporales, algunos pacientes con enfermedad ósea importante relacionada con aluminio tienen niveles séricos bajos de aluminio debido al depósito de éste en los tejidos blandos y el hueso.
En la prueba de desferroxamina, ésta se administra en una dosis de 40 mg/kg por vía I.V. durante un periodo de 30 minutos después de la diálisis. Los niveles basales del aluminio se comparan con los niveles máximos que aparecen 48 horas después de la administración de desferroxamina. La prueba se considera indicativa de toxicidad por aluminio cuando la diferencia entre las dos cifras supera los 200 µg/L.60 La biopsia ósea puede ayudar a confirmar el diagnóstico de enfermedad ósea relacionada con el aluminio; se observa con frecuencia osteomalacia con zonas extensas de material osteoide. La velocidad de la formación ósea, detectada por tetraciclina marcada, está disminuida y aparecen depósitos de aluminio a lo largo de la zona de calcificación principal.55
Al parecer, el hiperparatiroidismo protege contra la enfermedad ósea inducida por el aluminio. De hecho, se observa frecuentemente un agravamiento de la enfermedad en los pacientes después de la paratiroidectomía.56 Por lo tanto, debe quedar clara la naturaleza de la enfermedad ósea mediante biopsias óseas, y debe tratarse la toxicidad por aluminio.
Es especialmente importante definir el tipo de enfermedad ósea en vista de la menor frecuencia actual de la enfermedad relacionada al aluminio y al surgimiento de una nueva forma de osteodistrofia renal, la enfermeda ósea adinámica (aplásica). Esta es una lesión de recambio lento que se presenta en pacientes sin evidencia de acúmulo de aluminio u osteoide.58,61,62
La patogenia de la enfermedad ósea adinámica está mal comprendida. La enfermedad es más frecuente en pacientes con enfermedad renal terminal que reciben diálisis y que no tienen hiperparatiroidismo secundario (después de la paratiroidectomía), que han sido tratados con calcio y vitamina D o que tienen diabetes mellitus o intoxicación previa por aluminio.55,58,62,63 Se ha descrito asociación entre la diálisis peritoneal continua ambulatoria y la enfermedad ósea adinámica, que puede atribuirse a una mayor eficacia de la transferencia de calcio del dializado peritoneal al paciente, con supresión de la secreción de PTH en esta forma de diálisis.64 La presencia de enfermedad ósea adinámica en pacientes con función paratiroidea normal sugiere mayor producción de un inhibidor de la formación ósea o que no se producen factores de crecimiento para la formación ósea. También indica que puede requerirse de hipersecreción de PTH para mantener tasas normales de formación ósea en pacientes con enfermedad renal terminal y que la necesidad de mantener la remodelación ósea a un nivel normal puede ser un estímulo inherente para el hiperparatiroidismo en estos pacientes.55,61
Tratamiento
El tratamiento de la osteodistrofia renal debe dirigirse en primer lugar a evitar las complicaciones esqueléticas [ver tabla 4]. La restricción de fosfatos es crucial para mantener los niveles normales de calcio sérico y minimizar el desarrollo del hiperparatiroidismo secundario.54-56,59 Este control es importante, puesto que el aumento de los niveles de fosfato sérico puede aumentar el riesgo de calcificaciones metastásicas si las concentraciones de calcio sérico son normales (ver antes). Los niveles de fosfato sérico deben mantenerse entre 4 y 6 mg/dl.63
|
|
1,25-(OH)2D3- 1,25-dihidroxivitamina D3 DHT- dihidrotaquisterol PTH-hormona paratiroidea |
Se debe intentar reducir el consumo dietético de fosfatos con una dieta de 800-1,000 mg/día. Por desgracia, tal restricción no es muy aceptada, y en su lugar se pueden administrar antiácidos para unirse a los fosfatos en el intestino. Debido a que los antiácidos contienen magnesio, pueden producir hipermagnesemia en los individuos incapaces de excretar esta carga de magnesio; los antiácidos que contienen aluminio se han usado hasta hace poco como tratamiento estándar. Además de que estos compuestos producen un intenso estreñimiento, actualmente se sabe que inducen una absorción importante de aluminio.63 En consecuencia, la administración de aluminio como quelante de fosfatos debe ser mínima, sobre todo en los niños. Diversos investigadores son partidarios del uso de complementos de calcio (carbonato de calcio y acetato de calcio, pero no de citrato de calcio) como quelantes de fosfatos para evitar la toxicidad por aluminio.54-56,59,63 Se puede comenzar con dosis de 1 a 2 g/día, controlando el tratamiento mediante los niveles de fosfato en sangre. Normalmente, los complementos de calcio para corregir la hipocalcemia se administran lo más separado posible de las comidas. Sin embargo, cuando se usan para quelar fosfatos se deben administrar con las comidas para elevar al máximo el grado de fijación de fosfatos y evitar la hipercalcemia.55 Los niveles de calcio deben vigilarse estrechamente. Del creciente interés por la toxicidad del aluminio han surgido recomendaciones para el uso de menores concentraciones de calcio en las soluciones peritoneales y de hemodiálisis. Tales acciones previenen el desarrollo de hipercalcemia y permiten la administración de dosis mayores de calcio complementario por vía oral.55
Los análogos de la vitamina D también tienen un papel importante en el tratamiento de la osteodistrofia renal.55,59 Los pacientes con insuficiencia renal crónica pueden ser resistentes a las dosis habituales de vitamina D3 (400 U/día). Por lo tanto, la administración de 1,25-(OH)2D3 en dosis de 0.25 a 1.0 µg/día por vía oral o dehidrotaquisterol, un análogo sintético que no requiere de hidroxilación renal, en dosis de 0.2 mg/día puede aumentar la absorción intestinal de calcio, aumentando sus niveles séricos.
Existen cada vez más datos de que el 1,25-(OH)2D3 por vía intravenosa actúa a nivel del ARN mensajero, inhibiendo directamente la secreción de PTH, independientemente del efecto sobre los niveles séricos de calcio.65 Los estudios sugieren que la administración de 1,25-(OH)2D3 en pulsos por vía intravenosa tiene un efecto superior a la administración continua por vía oral en el tratamiento del hiperparatiroidismo secundario en los pacientes con hipercalcemia por la administración oral de vitamina D.66 La administración en pulsos produce una importante disminución en los niveles de PTH y fosfatasa alcalina en los pacientes en diálisis, así como una mejoría de la osteítis fibrosa, corroborada desde el punto de vista histológico, sin hipercalcemia concomitante.66
Debido a que la vitamina D aumenta la absorción intestinal de fosfato,56 es importante llevar un control de los niveles séricos de fosfato antes de instituir el tratamiento con vitamina D; de otro modo pueden presentarse calcificaciones metastásicas.
Si todos los métodos conservadores fracasan en el control del hiperparatiroidismo secundario y la hipercalcemia persiste (v.gr., si las concentraciones de calcio permanecen > 12 mg/dl) después de suspender el tratamiento con complementos de calcio y vitamina D y si en la biopsia ósea no se demuestran datos de toxicidad por aluminio, se debe considerar entonces la paratiroidectomía subtotal para el tratamiento de la osteítis fibrosa severa, las calcificaciones metastásicas o el prurito intratable.54-56
Si se demuestra toxicidad por aluminio con la prueba de desferoxamina57 y se confirma mediante biopsia ósea, el tratamiento quelante con desferoxamina debe mejorar los síntomas y los datos histológicos de la osteomalacia inducida por aluminio.55
El tratamiento de la enfermedad ósea adinámica sigue siendo un enigma. Sin embargo, la enfermedad puede relacionarse con la supresión excesiva de PTH por el calcio y la vitamina D. Es claro que aún se requiere mucho para saber más de esta nueva entidad.55,61,62
ANEMIA
Patogenia
La anemia ha sido reconocida desde hace mucho tiempo como una complicación de la insuficiencia renal crónica y puede además ser la causa de muchos de los síntomas relacionados con el síndrome urémico, como la fatiga, la debilidad y la reducción de la tolerancia al ejercicio.
La evaluación hematológica de los pacientes con insuficiencia renal crónica muestra anemia normocítica normocrómica en el frotis de sangre periférica.67 En ocasiones se observan células en erizo o en casco. Los niveles de hierro sérico, transferrina y ferritina son generalmente normales, salvo que se presenten hemorragias digestivas. En los pacientes con insuficiencia renal avanzada la vida media de los eritrocitos puede estar acortada.67 Algunos investigadores han sugerido que las toxinas presentes en el suero urémico pueden actuar como inhibidores de la producción eritroide o de su función. Las poliaminas espermina y espermidina han sido implicadas como toxinas.68 Sin embargo, estas moléculas parecen ser inhibidores generales de la hematopoyesis, más que inhibidores específicos de la eritropoyesis.67 La PTH también ha sido considerada como supresora de la eritropoyesis. Aunque la paratiroidectomía subtotal algunas veces puede producir aumento en la cantidad de los eritrocitos, la PTH no parece tener un efecto tóxico directo sobre la producción de eritrocitos; más bien, el exceso de PTH parece contribuir a la anemia al remplazar la médula ósea por tejido fibroso (osteítis fibrosa), limitando la producción celular de la serie eritroide.69
Actualmente está claro que la anemia de la insuficiencia renal crónica se debe principalmente a la deficiencia en la producción renal de eritropoyetina. Los niveles séricos de eritropoyetina en los pacientes urémicos suelen estar disminuidos de forma inadecuada para el grado de enemia.67 Además, el tratamiento sustitutivo con eritropoyetina humana recombinante corrige la anemia.70
Tratamiento
La eritropoyetina, uno de los factores estimuladores de colonias, es una glucoproteína que actúa como el mejor regulador de la producción de eritrocitos, y se libera en respuesta a la anoxia.70Al parecer estimula a la célula progenitora de la serie eritroide para diferenciarse en normoblastos, y también aumenta el reservorio de células formadoras de colonias. Aunque el hígado fetal es capaz de producir eritropoyetina, el riñón produce más del 90 porciento en los adultos. Las células endoteliales de los túbulos proximales o de los capilares peritubulares son considerados como los principales sitios de producción.70
Muchos estudios han confirmado que la eritropoyetina humana recombinante puede corregir la anemia en la insuficiencia renal crónica en etapas terminales y elimina virtualmente la necesidad de transfusiones en los pacientes que requieren diálisis.70-72 Se administra por vía intravenosa o subcutánea en dosis de 50 a 100 U/kg tres veces a la semana hasta llegar a una cifra de hematocrito de 30 a 35. El fármaco parece tener una toxicidad mínima y no se han detectado anticuerpos contra la eritropoyetina humana recombinante. Debido al aumento en la utilización del hierro, estimulado por la eritropoyesis, los depósitos de hierro deben remplazarse, vigilándose en forma estrecha los niveles de hierro, capacidad de fijación de hierro y ferretina.70-72 En la mayoría de los pacientes los suplementos de hierro por vía oral son suficientes (sulfato o gluconato ferroso, 325 mg o más, según tolerancia, tres veces al día). Sin embargo, si los niveles de ferritina son menores a 100 mg/ml se requiere tratamiento con hierro por vía parenteral.73
No sólo se ha demostrado objetivamente la eficacia de la eritropoyetina humana recombinante, sino que también han sido notables la mejoría subjetiva de los síntomas y de la calidad de vida.74 Los pacientes presentan una mejoría general notable, se sienten bien, mejoran su apetito, energía, libido, concentración y nivel de actividad.74,75 La anemia puede contribuir mucho más al síndrome urémico de lo que se pensaba, y su corrección ayuda en la rehabilitación de los pacientes con nefropatías en estado terminal.
La eritropoyetina humana recombinante también puede corregir la anemia y disminuye la necesidad de las transfusiones en los pacientes con insuficiencia renal moderada;76,77 esto reduce los riesgos de infecciones virales asociadas a transfusión (v.gr., citomegalovirus, hepatitis B y VIH) y la posible sensibilización de los candidatos a trasplantes.
El desarrollo de hipertensión o la exacerbación de la hipertensión previa, que requiere tratamiento antihipertensivo, se ha reconocido como un efecto colateral del uso de eritropoyetina en los pacientes en diálisis71,72 o que se van a someter a diálisis.76-78 Otra importante consideración es el efecto de la eritropoyetina en la evolución de la nefropatía moderada. Aunque estudios en animales han sugerido que el tratamiento de la anemia de la insuficiencia renal crónica con eritropoyetina puede exacerbar la velocidad de deterioro de la función renal,79,80 datos de estudios clínicos indican que no ocurre deterioro más rápido cuando se controla bien la presión arterial.76-78
TRASTORNOS HEMORRAGICOS POR UREMIA
Patogenia
Es bien conocido que los pacientes con insuficiencia renal crónica tienden a sangrar a pesar de tener tiempos de protrombina y tromboplastina parcial y un número de plaquetas normales. Los episodios hemorrágicos siguen siendo un factor importante que contribuye a la morbimortalidad relacionada con la uremia.81,82 Aunque la hemorragia de la mucosa gastrointestinal es la manifestación más frecuente de hemorragia urémica, también se presenta epistaxis, pericarditis hemorrágica y hematomas subdurales. La prueba de laboratorio que mejor se correlaciona con la tendencia a la hemorragia es el tiempo de sangrado.83
Aunque la naturaleza de la tendencia hemorrágica de la uremia continúa siendo difícil de describir, se han identificado alteraciones hemostásicas que incluyen defectos en la producción del factor 3 plaquetario, agregación plaquetaria subnormal, aumento en la producción de prostaciclinas y la liberación de un complejo anormal del Factor VIII-von Willebrand (FvW) por el endotelio vascular.67
Tratamiento
Varias medidas terapéuticas se han utilizado para el tratamiento de la hemorragia urémica [ver tabla 5].
|
||||||||||||||||||||
|
Diálisis Se ha sugerido que las alteraciones en la agregación plaquetaria pueden estar mediadas por moléculas que se encuentran en el suero de los individuos urémicos y que inhiben la actividad del factor 3 plaquetario. Se ha implicado al ácido guanidinsuccínico, a diversos compuestos fenólicos y a la urea. Debido a que estas toxinas potenciales son dializables, tradicionalmente se ha recomendado la diálisis para mejorar la función plaquetaria en la uremia.67 Sin embargo, este tratamiento no reduce significativamente el tiempo de sangrado ni las hemorragias clínicas en los pacientes urémicos. De hecho, la hemodiálisis puede aumentar el riesgo de hemorragia si se requieren anticoagulantes sistémicos para evitar la formación de trombos por el dializador.
Crioprecipitados Los crioprecipitados son concentrados multiméricos del complejo Factor VIII-FvW y se considera que el trastorno funcional de este complejo en la uremia explica la prolongación del tiempo de sangrado y la tendencia a las hemorragias. Las infusiones de crioprecipitados acortan el tiempo de sangrado y ayudan a controlar los episodios de hemorragias clínicas durante 12 a 18 horas, y los efectos son reproducibles.84 Las principales desventajas del tratamiento con crioprecipitados son la posibilidad de trasmisión de agentes infecciosos como el virus de la hepatitis y el VIH, además del costo para los pacientes.84
1-Desamino-8-D-arginina vasopresina El principal avance en el tratamiento de la hemorragia urémica ha sido el uso de 1-desamino-8-arginina vasopresina (DDAVP).85 Este análogo de la hormona antidiurética se ha usado para tratar la enfermedad de von Willebrand, puesto que aumenta los niveles circulantes del complejo factor VIII-FvW y disminuye el tiempo de sangrado.67,85
En los pacientes con uremia, la infusión de DDAVP (0.3 a 0.4 µg/kg en 20 ml de solución salina, administrados durante un periodo de treinta minutos) reduce notablemente el tiempo de sangrado y ha sido útil sobre todo para la prevención de las hemorragias clínicas producidas cuando se utilizan procedimientos invasivos como la obtención de biopsias o en cirugías.
El efecto máximo de la DDAVP se observa una o dos horas después de la infusión y tiene una duración de seis a ocho horas.85 Por desgracia, los efectos de la DDAVP se atenúan con las dosis repetidas.
El mecanismo mediante el cual la DDAVP corrige el tiempo de sangrado en los pacientes urémicos no está claro. Aunque la infusión de DDAVP se relaciona con un aumento en los niveles del complejo factor VIII-FvW, por lo general se encuentran niveles normales o elevados en los pacientes urémicos. Por lo tanto, se ha sugerido que la DDAVP puede actuar aumentando o cambiando los receptores de membrana de las plaquetas a los cuales se unen estos complejos, o por la inducción de la aparición de formas multiméricas mayores que puedan ser más activas.85
Estrógenos conjugados Aunque tanto los crioprecipitados como la DDAVP pueden corregir el tiempo de sangrado y permitir que se lleven a cabo intervenciones quirúrgicas sin hemorragias excesivas en los pacientes con insuficiencia renal, los efectos de ambos compuestos duran sólo unas pocas horas. Varios estudios han propuesto que los estrógenos conjugados (0.6 mg/kg/día durante cinco días), pueden acortar el tiempo de sangrado y mejorar la diátesis hemorrágica durante periodos más largos.86,87 Los efectos benéficos de los estrógenos comienzan seis a 24 horas después de su administración y el efecto máximo se observa en cinco a siete días. El efecto de la infusión de estrógenos durante cinco días puede durar hasta 14 días.
Los estrógenos conjugados no parecen afectar los niveles circulantes del factor VIII-FvW o cambiar la estructura multimérica. El mecanismo de acción todavía no está bien aclarado.87
Eritropoyetina Durante las pruebas con eritropoyetina para el tratamiento de la anemia de los pacientes en diálisis se observó que el tiempo de sangrado se acortaba.88,89 Curiosamente, se había observado previamente que las transfusiones administradas a los pacientes urémicos aumentaban el hematocrito a niveles mayores del 30 porciento y también corregían el tiempo de sangrado.89 No está claro si el efecto de la eritropoyetina es similar al observado con las transfusiones, pero el acortamiento del tiempo de sangrado puede ser un beneficio adicional de la eritropoyetina humana recombinante. El uso de la eritropoyetina para el control de las hemorragias también evita los riesgos de las transfusiones.
MANIFESTACIONES NEUROLOGICAS
Los trastornos neurológicos pueden ser los primeros síntomas que se observan en los pacientes con insuficiencia renal progresiva. Los síntomas iniciales pueden ser leves y consisten en disminución del pensamiento abstracto y de la concentración, irritabilidad, e insomnio, pero conforme avanza la insuficiencia renal se presentan síntomas más importantes que incluyen disminución de los reflejos tendinosos profundos con clonus, asterixis, convulsiones y depresión de las funciones mentales, con obnulación progresiva hasta el estupor y el coma.90
La principal complicación potencialmente incapacitante de la insuficiencia renal crónica avanzada es el desarrollo de una polineuropatía distal simétrica de tipo mixto, sensorial y motora. Esta entidad puede presentarse como el llamado síndrome de las piernas inquietas (incapacidad para controlar la actividad de las extremidades inferiores), como parestesias o hiperestesias de los pies, secundaria a neuropatía sensorial, o como debilidad distal de las extremidades inferiores.90
Debido a que las alteraciones en las funciones mentales (encefalopatía urémica) y algunos aspectos de la neuropatía urémica pueden corregirse mediante la diálisis, la presencia de síntomas neurológicos es una indicación clara de diálisis. Las secuelas neurológicas observadas en los pacientes en diálisis, tales como síndromes de desequilibrio y demencia por toxicidad del aluminio se revisan en otra parte de la obra.
COMPLICACIONES CARDIOVASCULARES
Las complicaciones cardiovasculares continúan siendo las principales causas de morbimortalidad en los pacientes con insuficiencia renal crónica.
Hipertensión
Más del 80 porciento de los pacientes con enfermedad renal en etapa terminal padecen de hipertensión; este es el factor de riesgo más importante de la uremia y contribuye a la insuficiencia renal cardiaca, la enfermedad cardiovascular y a la coronopatía observadas.91 La hipertensión no controlada acelera el descenso de la VFG. Dos mecanismos indudables contribuyen a la patogenia de la hipertensión en la insuficiencia renal: 1) la expansión de volumen por la notable retención de sal y agua, y 2) la activación del sistema renina-angiotensina-aldosterona.91 Ambos mecanismos pueden estar presentes en un mismo paciente, pero la diferenciación entre estos mecanismos patogénicos es útil en la clínica.
Cerca del 30 al 40 porciento de los pacientes urémicos, en particular los que están en tratamiento con diálisis, pueden lograr un control excelente de la presión arterial por medio de la eliminación del exceso de líquido con diuréticos o diálisis para lograr el llamado peso seco, con lo que se disminuye el uso de agentes hipertensivos. En comparación, en los pacientes que parecen tener como base un mecanismo dependiente de renina, la hipertensión requiere con frecuencia de dosis altas de agentes antihipertensivos a pesar de los intentos rigurosos para la eliminación de los líquidos mediante la diuresis o la diálisis.
Los inhibidores de la ECA deben usarse con precaución en los pacientes con estenosis bilateral de la arteria renal, con arteria única congénita o con estenosis unilateral del riñón trasplantado.92,93 En esta situación, la VFG depende considerablemente del aumento en la vasoconstricción arteriolar eferente mediada por la angiotensina II. Cuando se administran inhibidores de la ECA se presenta vasodilatación arteriolar, lo que causa disminución de la VFG y deterioro agudo de la función renal. Aún más, el potasio debe vigilarse con cuidado en los pacientes que reciben inhibidores de la ECA. Estos medicamentos están contraindicados en pacientes embarazadas.
Enfermedad pericárdica
La pericarditis urémica fue descrita inicialmente por Bright en su monografía clásica sobre la uremia en 1836.94 Los pacientes presentan de forma característica malestar torácico, frote pericárdico que se puede detectar en la exploración física, y con frecuencia arritmia auricular. El diagnóstico se hace mediante ultrasonografía cardiaca, que permite calcular el volumen del líquido pericárdico de forma no invasiva y sus efectos sobre la contractilidad del miocardio. Aunque la mayor parte de los pacientes con pericarditis tienen una pequeña cantidad de líquido en la parte posterior, la presencia de un derrame moderado en la parte anterior sugiere taponamiento cardiaco (ver adelante).
Tratamiento La presencia de pericarditis urémica es una indicación para iniciar la diálisis [ver figura 5]; la respuesta a este tratamiento ha sugerido que las toxinas urémicas dializables son factores patogénicos importantes.91 Se puede presentar hemopericardio, en particular cuando los pacientes reciben anticoagulantes sistémicos; por lo tanto, no debe usarse heparina en la hemodiálisis, o bien debe realizarse diálisis peritoneal.
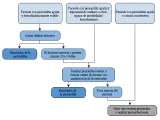
|
| Figura 5 |
| Tratamiento de la pericarditis en la insuficiencia renal crónica |
El tamaño del volumen pericárdico debe vigilarse en forma estrecha por medio de ultrasonido cardiaco. Se debe realizar punción y drenaje si se desarrolla inestabilidad hemodinámica o si el derrame es importante después de un tratamiento agresivo con diálisis durante un periodo de dos semanas (ver adelante).95,97 El desarrollo de hipotensión que no responde a la administración de líquidos durante la hemodiálisis es un dato importante que señala la presencia de taponamiento cardiaco.
Los pacientes también pueden desarrollar pericarditis después de la diálisis; esta complicación suele ocurrir en los casos de cirugía mayor, infecciones o en la diálisis inadecuada. En este tipo de pacientes la diálisis intensiva sólo es eficaz en el 60 por ciento de los casos y con frecuencia es necesario un método más agresivo, como el drenaje pericárdico.95-97
Los agentes antinflamatorios no esteroides pueden proporcionar alivio sintomático del dolor y la fiebre asociadas con pericarditis aguda relacionada a la diálisis, pero un estudio aleatorio, prospectivo y doble ciego mostró que la indometacina no afecta la historia natural de este padecimiento.96
De igual modo, aunque los esteroides sistémicos pueden producir alivio sintomático, no tienen efecto sobre el derrame, el taponamiento o el desarrollo de pericarditis constrictiva.97
El taponamiento pericárdico agudo es la complicación más grave de la pericarditis, y pone en peligro la vida. Por lo tanto, está indicado el drenaje en los pacientes que no responden al tratamiento intensivo con diálisis durante un periodo de dos semanas y si hay evidencias de que el derrame anterior persiste o se expande. Se debe colocar una sonda pericárdica percutánea que permita el drenaje y la instilación de esteroides no absorbibles, como acetato de triamcinolona (100 mg cada ocho horas durante 48 horas).98 Aunque la sonda puede ocasionar complicaciones de tipo técnico, suele tolerarse bien y es muy eficaz. En los casos en los que el paciente no responde a este tratamiento debe realizarse un drenaje quirúrgico abierto con la creación de una ventana pericárdica o una pericardiectomía.95-97
- Hunsicker LG, Levey AS: Progression of chronic renal disease: mechanisms, risk factors, and testing of interventions. The PrincipIes and Practice of Nephrology, 2nd ed. Jacobson HR, striker GE, Klahr S, Eds. Mosby-Year Book, St. Louis, 1995, p 622
- Brenner BM, Meyer TW , Hostetter TH. Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med 307:652, 1982
- Yamamoto T, Noble NA, Miller DE, et al: Sustained expression of TGF-beta 1 underlies development of progressive kidney fibrosis. Kidney Int 45:916, 1994
- Couser WG, Johnson RJ: Mechanisms of progressive renal disease in glomerulonephritis. Am J Kidney Dis 23:193, 1994
- Border W A, Noble NA: Cytokines in kidney disease: the role of transforming growth factor-beta. Am J Kidney Dis 22:105, 1993
- Klahr S, Ishidoya S, Morrissey J: Role of angiotensin II in the tubulointerstitial fibrosis of obstructive nephropathy. Am J Kidney Dis 26:141, 1995
- Brazy PC, stead WW , Fitzwilliam JF: Progression of renal insufficiency: role of blood pressure. Kidney Int 35:670, 1989
- Anderson S, Levey AS: Hypertension and progressive renal disease. The PrincipIes and Practice of Nephrology , 2nd ed. Jacobson HR, Striker GE, Klahr S, Eds. Mosby-Year Book, St. Louis, 1995, p 632
- Freedman BI, Iskandar SS, Appel RG: The link between hypertension and nephrosclerosis. Am J Kidney Dis 25:207, 1995
- Weisstuch JM, Dworkin LD: Does essential hypertension cause endstage renal disease? Kidney Int 36(suppl):S33, 1992
- Anderson S, Rennke HG, Brenner BM: Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 77:1993, 1986
- Lewis EJ, Hunsicker LG, Bain RP , et al: The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med 329:1456, 1993
- Hannedouche T, Landais P, Goldfarb B, et al: Randomised controlled trial of enalapril and beta blockers in non-diabetic chronic renal failure. BMJ 309:833, 1994
- Dworkin LD, Benstein JA, Parker M, et al: Calcium antagonists and converting enzyme inhibitors reduce renal injury by different mechanisms. Kidney Int 43:808, 1993
- Brown SA, Walton CL, Crawford P, et al: Long-term effects of antihypertensive regimens on renal hemodynamics and proteinuria. Kidney Int 43:1210, 1993
- Keane WF, Levey AS: Effects of diet and lipid-lowering agents on the progression of renal disease. The Principles and Practice of Nephrology , 2nd ed. Jacobson HR, Striker GE, Klahr S, Eds. Mosby-Year Book, St. Louis, 1995, p 640
- Klahr S, Buerkert J, Purkerson ML: Role of dietary factors in the progression of chronic renal disease. Kidney Int 24:579, 1983
- Okuda S, Nakamura T, Yamamoto T, et al: Dietary protein restriction rapidly reduces transforming growth factor-beta 1 expression in experimental glomerulonephritis. Proc Natl Acad Sci USA 88:9765, 1991
- Ihle BU, Becker GJ, Whitworth JA, et al: The effect of protein restriction on the progression of renal insufficiency. N Engl J Med 321:1773, 1989
- Zeller K, Whittaker E, Sullivan L, et al: Effect of restricting dietary protein on the progression of renal failure in patients with insulin-depen-dent diabetes mellitus. N Engl J Med 324:78, 1991
- Locatelli F, Alberti D, Graziani G, et al: Prospective, randomised, multicentre trial of effect of protein restriction on progression of chronic renal insufficiency. Lancet 337:1299, 1991
- Evanoff G, Thompson C, Brown J, et al: Prolonged dietary protein restriction in diabetic nephropathy. Arch Intern Med 149:1129, 1989
- Anderson S: Low protein diets and diabetic nephropathy. Semin NephroI 10:287, 1990
- Klahr S, Levey AS, Beck GJ, et al: The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. N Engl J Med 330:877, 1994
- Lumlertgul D, Burke TJ, Gillum DM, et al: Phosphate depletion arrests progression of chronic renal failure independent of protein intake. Kid-ney Int 29:658, 1986
- Harris DC, Falk SA, Conger JD, et al: Phosphate restriction reduces proteinuria of the uninephrectomized, diabetic rat. Am J Kidney Dis 11:489, 1988
- The Diabetes Control and Complications Trial Research Group: The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977, 1993
- Ketteler M, Noble NA, Border W A: Increased expression of transforming growth factor-beta in renal disease. Curr Opin Nephrol Hypertens 3:446, 1994
- Diamond JR: Macrophages and progressive renal disease in experimental hydronephrosis. Am J Kidney Dis 26:133, 1995
- Border WA, Noble NA: Mechanism of disease: transforming growth factor-beta in tissue fibrosis. N Engl J Med 331:1286, 1994
- Gibbons GH, Pratt RE, Dzau VJ: Vascular smooth muscle cell hypertrophy vs hyperplasia: autoimmune transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest 90:456, 1992
- Throckmorton D, Brogdon AP, Min B, et al: PDGF and TGF-B mediate collagen production by mesangial cells exposed to advanced glycosylation end products. Kidney Int 48:111, 1995
- Gesualdo L, Di Paolo S, Milani S, et al: Expression of platelet-derived growth factor receptors in normal and diseased human kidney: an immunohistochemistry and in situ hybridization study. J Clin Invest 94:50, 1994
- Feld S, Hirschberg R, Artishevsky A: Insulin-like growth factor I induces mesangial proliferation and increases mRNA and secretion of collagen. Kidney Int 48:45, 1995
- Fine LG, Kurtz I, Woolf AS, et al: Pathophysiology and nephron adaptation in chronic renal failure. Diseases of the Kidney, 5th ed., Vol 3. Schrier RW,Gottschalk CW, Eds. Little, Brown & Co, Boston, 1993, p 2703
- Meyer TW , Baboolal K, Brenner BM: Nephron adaptation to renal injury. The Kidney, 5th ed., Vol 2. Brenner BM, Ed. WB Saunders Co, Philadelphia, 1996, p 2011
- Bricker Ns, Fine LG, Kaplan M, et al: "Magnification phenomenon" in chronic renal disease. N Engl J Med 299:1287, 1978
- Rosa R, Battle D: Hyperkalemia. The Principies and Practice of Nephrology, 2nd ed. Jacobson HR, striker GE, KIahr S, Eds. Mosby Year Book, St. Louis, 1995, p 911
- Danovitch GM, Bourgoignie J, Bricker NS: Reversibility of the "salt-losing" tendency of chronic renal failure. N Engl J Med 296:14, 1977
- Levey AS, Perrone RD, Madias NE: Serum creatinine and renal function. Annu Rev Med 39:465, 1988
- Levey AS: Measurement of renal function in chronic renal disease. Kidney Int 38:167, 1990
- Mitch WE, Walser M, Buffington GA, et al: A simple method of estimating progression of chronic renal failure. Lancet 2:1326, 1976
- Ying CY, Tifft CP, Gavras H, et al: Renal revascularization in the azotemic hypertensive patient resistant to therapy. N Engl J Med 311:1070, 1984
- Jacobson HR, Striker GE: Report on a workshop to develop management recommendations for the prevention of progression in chronic renal disease. Am J Kidney Dis 25:103, 1995
- May RC, Mitch WE: Pathophysiology of uremia. The Kidney, 5th ed., Vol 2. Brenner BM, Ed. WB Saunders Co, Philadelphia, 1996, p 2148
- Mitch WE, Walser M. Nutritional therapy of the uremic patient. The Kidney, 5th ed., Vo 2. Brenner BM, Ed. WB Saunders Co, Philadelphia, 1996, p 2382
- Giovannetti S, Maggiore Q: A low nitrogen diet with proteins of high biologic value for severe chronic uremia. Lancet 1:1000, 1964
- Kopple JD: Dietary considerations in patients with advanced chronic renal failure, acute renal failure, and transplantation. Diseases of the Kidney, 5th ed., Vol 3. Schrier RW, Gottschalk CW, Eds. Little, Brown & Co, Boston, 1993, p 3167
- Giordano C: Use of exogenous and endogenous urea for protein synthesis in normal and uremic subjects. J Lab Clin Med 62:231, 1963
- May RC, Kelly RA, Mitch WE: Mechanisms for defects in muscle protein metabolism in rats with chronic uremia: influence of metabolic acidosis. J Clin Invest 79:1099, 1987
- Lefebvre A, de Vemejoul MC, Gueris J, et al: Optimal correction of acidosis changes progression of dialysis osteodystrophy. Kidney Int 36:1112, 1989
- Molitoris BA, Froment DH, Mackenzie TH, et al: Citrate: a major factor in the toxicity of orally administered aluminum compounds. Kidney Int 36:949, 1989
- Leaf A, Cotran RS: Renal pathophysiology. Chronic Renal Failure, 3rd ed. Oxford University Press, New York, 1985, p 191
- Andress DL, Sherrard DJ: The osteodystrophy of chronic renal failure. Diseases of the Kidney, 5th ed., Vol 3. Schrier RW, Gottschalk CW, Eds. Little, Brown & Co, Boston, 1993, p 2759
- Hruska KA, Teitelbaum SL: Renal osteodystrophy. N Engl J Med 333:166, 1995
- Cobum JW, Slatopolsky E: Vitamin D, parathyroid hormone, and the renal osteodystrophies. The Kidney, 4th ed., Vol 2. Brenner BM, Rector FC Jr, Eds. WB Saunders Co, Philadelphia, 1991, p 2036
- Nebeker HG, Coburn JW: Aluminum and renal osteodystrophy. Annu Rev Med 37:79, 1986
- Sherrard DJ, Hercz G, Pei Y, et al: The spectrum of bone disease in end- stage renal failure¾an evolving disorder. Kidney Int 43:436, 1993
- Malluche HH, Faugere MC. Renal bone disease 1990: an unmet challenge for the nephrologist. Kidney Int 38:193, 1990
- Milliner DS, Nebeker HG, Ott SM: Use of the deferoxamine infusion test in the diagnosis of aluminum-related osteodystrophy. Ann Inter Med 101:775, 1984
- Hercz G, Pei Y, Greenwood C, et al: Aplastic osteodystrophy without aluminum: the role of "suppressed" parathyroid function. Kidney Int 44:860, 1993
- Malluche HH, Monier-Faugere MC: Risk of adynamic bone disease in dialyzed patients. Kidney Int 38(suppl):S62, 1992
- Coburn JW, Salusky IB: Control of serum phosphorus in uremia. N Engl J Med 320:1140, 1989
- Coburn JW: Mineral metabolism and renal bone disease: effects of CAPD versus hemodialysis. Kidney Int 40(suppl):S92, 1993
- Delmez J, Tindira C; Groorns P, et al: Parathyroid hormone suppression by intravenous 1,25-dihydroxyvitamin D. J Clin Invest 83:1349, 1989
- Andress DL, Norris KC, Coburn JW , et al: Intravenous calcitriol in the treatrnent of refractory osteitis fibrosa of chronic renal failure. N Engl J Med 321:274, 1989
- Eschbach JW , Adamson JW: Hematologic consequences of renal failure. The Kidney, 4th ed., Vol 2. Brenner BM, Rector FC Jr, Eds. WB Saunders Co, Philadelphia, 1991. p 2019
- Segal GM, Stueve T, Adamson JW: Spermine and spermidine are nonspecific inhibitors of in vitro hematopoiesis. Kidney Int 31:72, 1987
- Barbour GL: Effect of parathyroidectomy on anemia in chronic renal failure. Arch Intern Med 139:889, 1979
- Eschbach JW: The anemia of chronic renal failure: pathophysiology and the effects of recombinant erythropoietin. Kidney Int 35:134, 1989
- Eschbach JW , Egrie JC, Downing MR, et al: Correction of the anemia of end-stage renal disease with recombinant human erythropoietin: results of a combined phase I and II clinical trial. N Engl J Med 316:73, 1987
- Nissenson AR, Nimer SD, Wolcott DL: Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy and nervous system effects. Ann Intern Med 114:402, 1991
- Van Wyck DB, Stivelman JC, Ruiz J. et al: Iron status in patients receiving erythropoietin for dialysis-associated anemia. Kidney Int 35:712, 1989
- Eschbach JW , Adamson JW : Recombinant human erythropoietin: implications for nephrology .Am J Kidney Dis 11:203, 1988
- Eschbach JW, Abdulhadi MH, Browne JK, et al: Recombinant human erythropoietin in anemic patients with end-stage renal disease: results of a phase III multicenter clinical trial. Ann Intern Med 111:992, 1989
- Eschbach JW, Kelly MR, Haley NR, et al: Treatment of the anemia of progressive renal failure with recombinant human erythropoietin. N Engl J Med 321:158, 1989
- The US Recombinant Human Erythropoietin Predialysis Study Group: Double-blind, placebo-controlled study of the therapeutic use of recombinant human erythropoietin for anemia associated with chronic renal failure in predialysis patients. Am J Kidney Dis 18:50, 1991
- Lim Vs, DeGowin RL, Zavala D, et al: Recombinant human erythropoietin treatment in pre-dialysis patients: a double-blind placebo-controlled trial. Ann Intern Med 110:108, 1989
- Garcia DL, Anderson S, Rennke HG, et al: Anemia lessens and its prevention with recombinant human erythropoietin worsens glomerular injury and hypertension in rats with reduced mass. Proc Natl Acad Sci USA 85:6142, 1988
- Lafferty HM, Anderson S, Brenner BM: Anemia: a potent modulator of renal hemodynamics in models of progressive renal disease. Am J Kidney Dis 17(suppl 1):2, 1991
- Carvalho ACA: Bleeding in uremia: a clinical challenge. N Engl J Med 308:38, 1983
- Deykin D: Uremic bleeding. Kidney Int 24:698, 1983
- Steiner RW, Coggins C, Carvalho AC: Bleeding time in uremia. a useful test to assess clinical bleeding. Am J HematoI 7:107, 1979
- Janson PA, Jubelirer SJ, Weinstein MJ, et al: Treatment of the bleeding tendency in uremia with cryoprecipitate. N Engl J Med 303:1318, 1980
- Mannucci PM, Remuzzi G, Pusineri F, et al: Deamino-8-D-arginine vasopressin shortens the bleeding time in uremia. N Engl J Med 308:8, 1983
- Liu YK, Kosfeld RE, Marcum SG: Treatment of uraemic bleeding with conjugated estrogen. Lancet 2:887, 1984
- Livio M, Mannucci PM, Vigano G, et al: Conjugated estrogens for the management of bleeding associated with renal failure. N Engl J Med 315:731, 1986
- Moia M, Mannucci PM, Vizzotto L, et al: lmprovement in the haemostatic defect of uraemia after treatment with recombinant human erythropoietin. Lancet 2:1227, 1987
- Vigano G, Benigni A, Mendogni D, et al: Recombinant human erythropoietin to correct uremic bleeding. Am J Kidney Dis 18:44, 1991
- Fraser CL, Arieff Al: Nervous system manifestations of renal failure. Diseases of the Kidney, 5th ed., Vol 3. Schrier RW, Gottschalk CW, Eds. Little, Brown & Co, Boston, 1993, p 2789
- Kim KE, Swartz C: Cardiovascular complications of end-stage renal disease. Diseases of the Kidney, 5th ed., Vol 3. Schrier RW, Gottschalk CW, Eds. Little, Brown & Co, Boston, 1993, p 2817
- Hricik DE, Browning PJ, Kopelman R, et al: Captopril-induced functional renal insufficiency in patients with bilateral renal-artery stenoses or renal artery stenosis in a solitary kidney. N Engl J Med 308:373, 1983
- Curtis JJ. Luke RG, Whelchel JD, et al: Inhibition of angiotensin-converting enzyme in renal-transplant recipients with hypertension. N Engl J Med 308:377, 1983
- Bright R: Tabular view of the morbid appearance in 100 cases connected with albuminous urine: with observations. Guys Hosp Rep 1:380, 1836
- Rutsky EA, Rostard SG: Pericarditis in end stage renal disease: clinical characteristics and management. Seminars in Dialysis 2:25, 1989
- Spector D, Alfred H, Siedleck M, et al: A controlled study of the effect of indomethacin in uremic pericarditis. Kidney Int 24:663, 1983
- Ventura SG, Garech GS: The management of pericardial disease in renal failure. Seminars in Dialysis 3:21, 1990
- Buselmeier TJ, Davin m, Simmons RL, et al: Treatment of intractable uremic pericardial effusion: avoidance of pericardiectomy with local steroid instillation. JAMA 240:1358, 1978