Oncología
⭳ Abrir artículo (PDF)803.7 KBEste artículo fue revisado respecto a la Edición 3/2000. Ver esa versión →
Contenido del artículo
II GENETICA MOLECULAR DEL CANCER
- El cáncer como una enfermedad genética
- Oncogenes y proto-oncogenes
- MUTACIONES QUE CAUSAN ACTIVACION O MODIFICACION EN LA FUNCION
- FUNCION DE LOS ONCOGENES IN VITRO Y EN MODELOS MURINOS
- Genes supresores de tumores
- REVERSION DE LA TUMORIGENICIDAD
- EL MODELO DE KNUDSON Y LAS MUTACIONES DE PERDIDA DE FUNCION
- PROGRESION DEL CICLO CELULAR Y VIA DEL RB1
- GENES DE ESTABILIDAD GENOMICA: p53 y BRCA1
- SUPRESORES DE TUMORES QUE PARTICIPAN EN LA SEÑALIZACION Y DIFERENCIACION
- Inestabilidad de microsatélites y reparación de errores en el ADN
- Progresión tumoral
- ACUMULO DE LESIONES GENETICAS
- MUERTE CELULAR E INMORTALIDAD
- METASTASIS Y ANGIOGENESIS
- RESISTENCIA A LOS FARMACOS QUIMIOTERAPICOS
- Implicaciones clínicas del análisis genético
II GENETICA MOLECULAR DEL CANCER
DR. DANIEL A. HABER, Ph.D.
El cáncer como una enfermedad genética
La expansión clonal no controlada de una célula, que con frecuencia causa invasión de los tejidos circundantes y diseminación metastásica, produce el cáncer. Las alteraciones genéticas iniciales en esta célula que desencadenan la proliferación aberrante son seguidas por acúmulo de mutaciones adicionales entre su progenie. Por último, ocurre un proceso de selección por el que las subclonas con propiedades de mayor crecimiento se vuelven dominantes dentro del tumor, la llamada progresión tumoral. En algunos tumores se ha definido una evolución genética histológica y molecular clara desde las lesiones precancerosas hasta el cáncer francamente maligno e invasor (v.gr., cánceres de cólon y vejiga). Sin embargo, en muchas neoplasias este proceso puede no ser clínicamente evidente. Los genes que están alterados en el desarrollo y progresión del cáncer aún se investigan en forma intensa. En muchos casos la identificación de estos genes ha contribuido a un mayor conocimiento de los mecanismos fisiológicos normales que controlan la proliferación celular. Los productos de estos genes participan en el control del ciclo celular básico, la trasmisión de señales de crecimiento, la regulación de la diferenciación y muerte celular, y en el establecimiento de la inmortalidad celular. Típicamente ocurre alteración de estos genes en las células somáticas destinadas a ser cancerosas. Sin embargo, en raros casos las mutaciones pueden trasmitirse en línea germinal, causando una predisposición genética al cáncer (i.e., síndromes familiares de cáncer).
Se piensa que los factores ambientales también contribuyen en el desarrollo del cáncer. Las interacciones entre factores ambientales y variaciones genéticas sutiles que distinguen a los individuos pueden constituir un determinante importante en el riesgo de cáncer en la población general. En algunos casos existe una relación directa entre el efecto del carcinógeno sobre el ADN y mutaciones específicas en el gen supresor de tumores p53 (v.gr., exposición a tabaco o a la toxina de hongos aflatoxina). En otros casos, efectos más indirectos pueden aumentar el riesgo de desarrollar cáncer (v.gr., exposición a carcinógenos potentes como asbesto o exposiciones más sutiles, como tratamiento prolongado con estrógenos). La exposición a la radiación ionizante es otra causa de cáncer. La incidencia de leucemias y de varios tumores sólidos entre los sobrevivientes a la bomba atómica proporcionó datos para estudios clásicos sobre las dosis de radiación y sus consecuencias. Datos más recientes se han obtenido a partir de casos de complicaciones tardías de radioterapia o quimioteraapia radiomimética administradas a pacientes como tratamiento para una neoplasia inicial.
Por último, las infecciones virales se han relacionado con el desarrollo de cánceres específicos. Estos incluyen el carcinoma cervical causado por subtipos específicos de papilomavirus humano, el carcinoma hepatocelular resultado de la infección crónica con virus de la hepatitis B, el carcinoma nasofaríngeo y los linfomas en huéspedes inmunosuprimidos que se asocian con virus Epstein-Barr, la rara leucemia aguda de células T causada por el virus linfotrópico transformador de células T humanas de tipo I y, potencialmente, el sarcoma de Kaposi que se asocia con herpesvirus humano tipo 8. Con estas excepciones, la mayoría de los cánceres en humanos no son causados por una infección viral. Sin embargo, mucho de nuestro conocimiento sobre los genes humanos que participan en el cáncer derivó originalmente de estudios de virus que provocan tumores en pollos y roedores. La alteración por estos virus de genes celulares que participan en la proliferación celular llevó a la identificación de los oncogenes, proporcionando la primera clave para los eventos genéticos que causan el cáncer en los humanos.
Oncogenes y proto-oncogenes
MUTACIONES QUE CAUSAN ACTIVACION O MODIFICACION EN LA FUNCION
Los genes que causan cáncer, u oncogenes, fueron descubiertos al encontrar que genes específicos de retrovirus de pollos y roedores eran capaces de transformar las células normales de mamíferos en cultivos. Se demostró que estos genes virales transformadores eran homólogos de los genes de mamíferos (proto-oncogenes) que habían sido robados de la célula huésped durante la evolución viral por su capacidad para estimular la proliferación celular.1 Los cánceres humanos primarios, aunque no son causados por una infección viral, tienen alelos activados similares de proto-oncogenes. Entre los primeros oncogenes descubiertos fueron los que codifican proteínas que participan en forma directa en la trasmisión de las señales de proliferación celular. Estos incluyen receptores para factores de crecimiento que están activados en forma constitutiva, aunque responden a la presencia continua de un factor de crecimiento (v.gr., receptor del factor de crecimiento derivado de plaquetas o receptor del factor de crecimiento epidérmico) así como a moléculas que le inhiben (i.e., moléculas que actúan sobre una región del ADN que se localiza en el lado 3´de un gen) que normalmente cambian entre el estado activo e inactivo pero que están mutadas en las células neoplásicas para mantenerse en una posición permanente de activación (v.gr., H-ras, K-ras y N-ras). Los mecanismos por los que estas proto-oncogenes celulares normales se activan en los cánceres humanos incluyen mutaciones puntiformes, amplificación de genes y traslocación de cromosomas [ver tabla 1 y figura 1]. Debido a que causan propiedades funcionales nuevas o alteradas en la proteína que codifican y son genéticamente dominantes sobre el segundo alelo normal, estas mutaciones comúnmente son conocidas como de modificación de función.
Mutaciones puntiformes
Las mutaciones puntiformes que son capaces de activar un producto genético son poco comunes y reflejan propiedades funcionales específicas de la proteína en sí. Por ejemplo, solo las alteraciones específicas en los tres codones de la familia de proteínas ras causan una activación constitutiva de la señal ras.2 Se supone que otras mutaciones son funcionalmente silenciosas o causan proteínas inactivas y por lo tanto no son seleccionadas durante la transformación neoplásica. En forma semejante, cambios específicos en los aminoácidos de receptores de factores de crecimiento u otras moléculas de señal confieren un efecto activo o inductor de crecimiento o evitan la inhibición por parte de las señales fisiológicas apropiadas. De especial interés son las mutaciones en el gen ret, un receptor de factor de crecimiento y el único proto-oncogén encontrado que muta en la línea germinal de pacientes predispuestos a cáncer. Las sustituciones de aminoácidos con diferentes dominios funcionales de la proteína causan neoplasia endócrina múltiple tipo IIA o tipo IIB o cáncer medular de tiroides, mientras que las mutaciones inactivas provocan un defecto en el desarrollo que afecta la innervación del colon, la enfermedad de Hirschsprung.3 La estrecha relación entre las diferentes mutaciones en el gen ret y diversos síndromes neoplásicos y de desarrollo parece deberse a diversas propiedades funcionales que son mediadas por los varios dominios de la proteína ret así como por diferentes vías asociadas a ret que pueden activarse en distintos tipos celulares.
Amplificación de genes y traslocaciones cromosómicas
Además de las sustituciones de aminoácidos que causan un producto genético activado, los proto-oncogenes pueden convertirse a sus formas oncogénicas por alteraciones cromosómicas grandes. La amplificación genética, que consiste en la sobrerreplicación de fragmentos grandes de cromosomas, es un mecanismo común para la expresión exagerada aberrante de genes específicos en los tumores.4 Los genes amplificados, en ocasiones hasta en cientos de copias de los genes diploides normales, pueden residir en elementos extracromosómicos pequeños (cromosomas doble punto) o pueden integrarse dentro de una sola localización cromosómica, detectables como una región grande de tinción homogénea. En las células neoplásicas la pérdida de los genes celulares que contribuye normalmente al mantenimiento de la integridad genómica (v.gr., p53) puede facilitar los eventos cromosómicos aberrantes, incluyendo amplificación genética y, por lo tanto, acumulación de lesiones genéticas adicionales. Entre los oncogenes mejor conocidos que son amplificados por células neoplásicas está N-myc. Este factor de transcripción, que tiene un papel fisiológico en la estimulación de la proliferación celular, comunmente está amplificado en el neuroblastoma pediátrico, en donde confiere un mal pronóstico clínico.5 La expresión exagerada de los proto-oncogenes puede causar también una traslocación o rearreglo cromosómico, que mueve el gen de su promotor regulado fisiológicamente y lo coloca bajo el control de un promotor extraño y más activo.6 Por ejemplo, en el linfoma de Burkitt el locus cromosómico que contiene al gen c-myc se rearregla de modo que se pierden las regiones reguladoras colocadas en dirección superior (i.e., la región localizada del lado 5´del gen); la expresión del gen es dirigida por el intenso promotor de las cadenas pesadas de las inmunoglobulinas, que activa en forma constitutiva a los linfocitos B.7,8 La desregulación de la expresión de c-myc en estas células proporciona un potente estímulo para la proliferación celular.
La primera traslocación cromosómica específica identificada en el cáncer humano fue el cromosoma Filadelfia, que define a la leucemia mieloide crónica (LMC). La fusión de los cromosomas 9 y 22 causa la unión de dos genes no relacionados, el gen c-abl que codifica una tirosina cinasa, localizado en el cromosoma 9, y el gen bcr (por recombinación de punto de ruptura) localizado en el cromosoma 22.9 Por este rearreglo cromosómico se forma una proteína quimérica con nuevas propiedades de transformación. Las variantes poco comunes de LMC, en las que el cromosoma Filadelfia no es evidente por análisis citogenético, pueden tener rearreglos genéticos complejos, causando una traslocación bcr-abl que puede detectarse solo con marcadores moleculares. La fase acelerada o blástica de la LMC suele asociarse con duplicación del cromosoma Filadelfia, lo que sugiere que el aumento de copias de este gen aberrante confiere un efecto transformador dependiente de dosis. La fusión bcr-abl también demuestra una especificidad marcada al tipo celular: la clásica ruptura cromosómica se asocia con proliferación mieloide, mientras que una ruptura variante, que causa una alteración sutil en la proteína quimérica, origina la leucemia linfoide pediátrica, que tiene mal pronóstico.10 La importancia de la traslocación bcr-abl consiste en que el evento genético inicial en la LMC está apoyada por un modelo murino, en el que la expresión transgénica de la proteína quimérica bcr-abl en las células hematopoyéticas es suficiente para desencadenar proliferación tanto mieloide como linfoide.11
Otras traslocaciones cromosómicas que generan proteínas quiméricas nuevas se han asociado con diversos tipos de leucemia.12 En la leucemia promielocítica aguda (LPA), un rearreglo cromosómico une un gen nuevo, PML, con el gen del receptor-alfa del ácido retinoico (RAR-alfa). Aunque se desconocen las propiedades funcionales precisas de la molécula quimérica PML-RAR-alfa, esta traslocación puede explicar la respuesta dramática de esta leucemia al tratamiento con ácido retinoico-trans, que ha revolucionado el cuidado clínico de los pacientes con LPA.13 También se ha identificado otro producto de traslocación cromosómica nuevo, el TEL-AML1, que ocurre en una forma común y con buena respuesta a tratamiento de leucemia linfoblástica aguda pediátrica.14
La capacidad de las células leucémicas para crecer en cultivo lo suficiente para realizar un análisis citogenético ha facilitado la caracterización de las traslocaciones cromosómicas en las leucemias. Sin embargo, también se han observado traslocaciones cromosómicas específicas en tumores sólidos. Destacan el sarcoma de Ewing y la familia de tumores neuroepiteliales periféricos (TNEP). Estos tumores antes mal definidos se sabe que se asocian con una traslocación cromosómica que fusiona el gen EWS con diversos factores de transcripción de la familia del gen ETS (la proteína quimérica más común es EWS-FLI1).15 Este producto quimérico parece actuar directamente sobre los promotores para dirigir la expresión de genes que inducen la proliferación celular. La identificación de las traslocaciones EWS permitió la clasificación molecular de una clase de tumores cuya proliferación se debe a alteraciones genéticas semejantes y que responde a esquemas quimioterápicos parecidos.
FUNCION DE LOS ONCOGENES IN VITRO Y EN MODELOS MURINOS
La detección de mutaciones de cambio de función en los proto-oncogenes proporciona evidencia intensa de que estas alteraciones genéticas contribuyen a la transformación maligna. Sin embargo, el tiempo en que ocurren estas mutaciones durante el inicio y la progresión del cáncer puede ser variable. Con la sola excepción de c-ret, no se observan mutaciones de los proto-oncogenes en la línea germinal de los pacientes con síndromes familiares de cáncer, quizá porque tienen consecuencias adversas en el desarrollo normal. Las traslocaciones cromosómicas parecen constituir el evento inicial para la transformación maligna en algunas leucemias y tumores sólidos. En otros cánceres, las mutaciones en los proto-oncogenes pueden contribuir a la progresión del cáncer una vez que ya se ha desarrollado. Dos modelos experimentales, los ratones transgénicos y el cultivo celular, han sido de gran valor para reconocer las propiedades funcionales de los oncogenes. Los ratones transgénicos son generados por la inyección de plásmidos de ADN recombinante en un huevo fertilizado.16 Estos plásmidos pueden contener un oncogén cuya expresión esté dirigida por un promotor tisular específico. Una fracción de la progenie del ratón demostrará la expresión esperada del constructo transgénico. Aunque la expresión de ciertos oncogenes en los tejidos apropiados puede causar un crecimiento maligno, en otros casos el cáncer se desarrolla solo después del apareamiento de ratones que expresan diferentes oncogenes. La expresión de dos oncogenes diferentes en la progenie puede ayudar a inducir el crecimiento maligno.17 También se ha probado la función de oncogenes dominantes por medio de la transformación de células de roedores in vitro. Mientras que las líneas celulares establecidas requieren de poco estímulo para volverse tumorigénicas, las células cultivadas, como las derivadas de riñón de rata bebé o de fibroblastos de embrión de ratón, se transforman solo después de la introducción de combinaciones de potentes oncogenes.18 Por ejemplo, la combinación de c-myc y H-ras activado es eficaz para transformar a las células primarias, pero ninguno de estos dos oncogenes es suficiente en forma aislada.
Además de su papel para validar la posibilidad de transformación de los proto-oncogenes activados, los ensayos funcionales sobre el estudio de los virus ADN tumorales han proporcionado conocimiento importante sobre la relación entre los oncogenes y otra clase de genes relacionados al cáncer, los supresores de tumores. A diferencia de los retrovirus que tienen versiones activadas de proto-oncogenes celulares incorporadas a sus propios genomas, los virus ADN tumorales, como el adenovirus, el SV40 y el papilomavirus, codifican proteínas virales originales capaces de transformar a las células de roedores, monos o humanos. La introducción del antígeno SV40 T a ratones transgénicos causa el desarrollo de tumores en prácticamente cualquier tejido en el que se exprese. En forma semejante, la combinación de las proteínas adenovirales E1A y E1B es altamente transformadora en ensayos de trasnformación celular primaria. Un descubrimiento importante respecto a la etiología del cáncer fue el hallazgo de que los genes transformadores derivados de estos tres virus ADN tumorales no relacionados tenían la misma función, que consiste en la inactivación de los genes celulares supresores de tumores p53 y RB1.19 Por lo tanto, el cáncer se origina tanto por aumento de señales de proliferación como por pérdida de genes que inhiben la proliferación celular.
Genes supresores de tumores
REVERSION DE LA TUMORIGENICIDAD
Antes conocidos como anti-oncogenes, los genes supresores de tumores constituyen una familia de genes celulares cuya inactivación durante la tumorigénesis promueve el crecimiento maligno [ver tabla 2]. Su descubrimiento inicial surgió de tres líneas convergentes de investigación: el análisis de las propiedades funcionales de las células normales contra las cancerígenas, estudios epidemiológicos de cáncer en niños y el análisis genético molecular de pérdidas cromosómicas en tumores. Uno de los descubrimientos iniciales fue que la fusión de una célula maligna a una maligna causaba una célula híbrida que perdía sus propiedades malignas [ver figura 2].20 Este dato inesperado indicaba que el estado de malignidad era recesivo y sugería que genes en la célula no maligna podrían restablecer el control del crecimiento normal de una célula maligna que aparentemente había perdido información genética. Estos experimentos fueron seguidos por la demostración de que cromosomas individuales aislados de células normales eran capaces de revertir la tumorigenicidad de las células neoplásicas.21 La clonación molecular de genes específicos supresores de tumores hizo posible después demostrar que los plásmidos de ADN recombinante que contenían estos genes eran capaces de suprimir el crecimiento tumoral.
EL MODELO DE KNUDSON Y LAS MUTACIONES DE PERDIDA DE FUNCION
La mayoría de los cánceres se originan durante la edad adulta; al parecer el aumento en la incidencia de cáncer al aumentar la edad es un reflejo del acúmulo del daño genético en las células tronco viejas. Los cánceres pediátricos, aunque raros, típicamente se desarrollan en células tronco jóvenes, como las células tronco renales que originan el tumor de Wilms, las células primitivas del neuroectodermo que se transforman en neuroblastoma y los retinoblastos que pueden producir el tumor ocular retinoblastoma. Un dato característico de estos tumores pediátricos es la existencia de familias en las que aproximadamente la mitad de los niños desarrollan tipos específicos de tumores. Dentro de estas familias con predisposición al cáncer los tumores que se originan con frecuencia son bilaterales o multicéntricos desde el principio y se desarrollan antes que lo correspondiente al comparar con los casos aislados en la población general. Al usar la distribución de Poisson para determinar la probabilidad de desarrollar cáncer como una función de la edad en los cánceres familiares comparado con los cánceres pediátricos esporádicos, Alfred Knudson propuso el modelo que forma ahora el fundamento de la genética del cáncer humano [ver figura 3].22
El modelo Knudson predice que los niños con tumores familiares han heredado una alteración genética inicial y requieren solo otra alteración genética adicional para iniciar la tumorigénesis. Por el contrario, los niños con tumores esporádicos necesitan adquirir dos alteraciones genéticas independientes dentro de la misma célula, un evento poco probable que explica porqué los cánceres esporádicos son menos frecuentes, de presentación unilateral y de inicio más tardío. Estudios genéticos subsecuentes en dos de los tumores estudiados por Knudson identificaron estos llamados golpes o alteraciones genéticas como la inactivación secuencial de los dos alelos de un gen supresor de tumores crítico: el RB1 en el retinoblastoma23 y el WT1 en el tumor de Wilms.24 El modelo Knudson explica también la paradoja respecto a que las mutaciones en genes supresores de tumores son de pérdida de función o recesivas [ver tabla 2], a pesar de que el cáncer familiar se presenta con un rasgo autosómico dominante. Aunque la pérdida de un solo alelo de un gen supresor de tumores puede ser funcionalmente silenciosa en presencia de un segundo alelo normal, la frecuencia de mutaciones espontáneas es suficientemente alta para asegurar que por lo menos una célula dentro de la célula blanco tenga posibilidad de perder un segundo alelo e iniciar la transformación maligna [ver figura 3].
Los genes supresores de tumores fueron identificados por la aplicación de herramientas citogenéticas y de genética molecular a estas observaciones funcionales y epidemiológicas. Mientras que la mutación inicial que inactiva un alelo de un gen supresor de tumor es típicamente una mutación puntiforme dentro de un gen en sí, la pérdida de un segundo alelo frecuentemente causa una deleción cromosómica o rearreglo mayor.25 La segunda alteración puede identificarse con facilidad por análisis de cariotipo, pero con más frecuencia se requiere análisis molecular para detectar la pérdida de marcadores polimórficos asociados en forma estrecha con el gen blanco. Los marcadores polimórficos (como los fragmentos de restricción de longitud polimórfica) son variables genéticas que pueden usarse para identificar cromosomas heredados de un padre [ver figura 4]. La pérdida de un polimorfismo derivado de cualquiera de los padres (llamada pérdida de la heterocigosidad [PH]) es indicación de que un gen supresor de tumores vecino ha sido inactivado en un cáncer. Estas pérdidas alélicas, que pueden mapearse en todos los cromosomas para un tipo determinado de tumor, han sido más eficaces para aislar genes supresores de tumores individuales, típicamente con estrategias de clonación posicional.
PROGRESION DEL CICLO CELULAR Y VIA DEL RB1
El primer supresor de tumores identificado fue el gen RB1, que participa en el tumor ocular pediátrico retinoblastoma.23 De muchas maneras, el RB1 sigue siendo el prototipo de esta clase de genes y su conexión íntima con el mecanismo básico de progresión del ciclo celular ilustra la estrecha relación entre la proliferación celular normal y la transformación maligna. Como lo predice el modelo Knudson,22 un alelo de RB1 es inactivado en la línea germinal de niños con retinoblastoma familiar. Sus tumores demuestran PH, lo que indica pérdida somática del alelo restante. Los niños con retinoblastoma esporádico, que constituyen el 90 por ciento de los casos, tienen ambos alelos RB1 inactivados dentro de una sola célula somática. Los niños que portan una mutación en un alelo RB1 también tienen predisposición a osteosarcomas, aunque no tienen mayor susceptibilidad a otros tipos de cáncer en los que se ha demostrado pérdida somática del RB1 (v.gr., cáncer de pulmón). Esto ilustra una paradoja importante en la genética del cáncer: un gen expresado en todos los tejidos normales controla el inicio del cáncer solo en órganos seleccionados, en otros parece ser solo una más de muchas alteraciones genéticas que contribuyen al estado de neoplasia. Las explicaciones actuales para esta paradoja incluyen diferencias potenciales en el ensamble genético de diferentes tipos célulares, así como diferencias en los mecanismos compensatorios que pueden activarse después de la pérdida de un gen supresor de tumores (v.gr., el desencadenamiento de la muerte celular).
La proteína codificada por RB1 es una fosfoproteína nuclear que se une a productos de una familia de genes llamada E2F, que a su vez se asocian con proteínas de la familia DP.26 Los complejos E2F-DP tienen un papel crucial al activar la transcripción de los genes requeridos para la síntesis de ADN. La unión a RB1 suprime este efecto, esencialmente al bloquear a las células en la fase G1 del ciclo celular. Durante la proliferación celular normal, un complejo de cinasa que regula el ciclo celular y que contiene ciclina D y cinasa 4 dependiente de ciclina (DDK4) fosforila a RB1, causando su liberación del complejo E2F-DP, lo que permite que la célula entre a la fase de síntesis (S) del ADN del ciclo celular. Aunque la inactivación fisiológica de la RB1 a través de la hiperfosforilación es transitoria y reversible, la inactivación de RB1 por mutación, común en muchos tipos diferentes de tumores, permite un estímulo constante para la proliferación celular. En forma semejante, los virus ADN oncogénicos codifican proteínas que inactivan de modo específico e irreversible al producto del gen RB1, como la proteína E1A del adenovirus, el antígeno T del SV40 y la proteína E7 del papilomavirus. El RB1 en sí es un miembro de una familia de genes, con dos genes muy relacionados denominados p107 y p130. Aún no se sabe por qué el RB1 es un blanco frecuente de mutaciones en los cánceres humanos mientras que estos genes similares no mutan. El mejor conocimiento en las diferencias sutiles de la función de estos otros genes podrá explicar este hecho.
El estudio sobre la regulación normal del RB1 ha proporcionado también conocimientos importantes sobre otros genes relacionados con el cáncer que participan en la regulación del ciclo celular [ver figura 5]. El ciclin D, un gen cuya expresión causa hiperfosforilación e inactivación de RB1, suele amplificarse y expresarse en forma exagerada en el cáncer de mama. Por el contrario, p16/INK4a, un inhibidor de CDK4 que favorece la forma activa hiperfosforilada de RB1, sufre deleción en gran variedad de tumores. Las mutaciones de línea germinal de p16/INK4a son causa del melanoma familiar.22 Por último, también se han demostrado mutaciones en el gen cinasa CDK4 que hacen que la proteína codificada sea insensible a la inhibición por p16/INK4a, favoreciendo la inactivación de RB1, en cánceres humanos y en líneas de melanoma. Un dato característico de las mutaciones en estos diferentes componentes del ciclo celular es que parecen ser mutuamente excluyentes. Un solo tumor tiene una alteración en solo uno de estos genes, lo que es evidencia de que estas mutaciones son funcionalmente equivalentes y que no se confiere ninguna mayor ventaja de crecimiento por el acúmulo de mutaciones adicionales en componentes de la misma vía.28
GENES DE ESTABILIDAD GENOMICA: p53 y BRCA1
Aunque el gen supresor de tumores RB1 es un componente central de la regulación del ciclo celular, p53 tiene un papel crucial en el mantenimiento de la integridad genómica, de ahí su designación popular como el "guardián del genoma".29 p53 se expresa normalmente en niveles bajos en todas las células. Sin embargo, las lesiones genéticas, como la radiación ionizante, inician la estabilización y activación de la proteína p53. La p53 funciona como un factor de transcripción, dirigiendo la expresión de p21, un inhibidor de cinasas dependiente de ciclinas que regula el ciclo celular [ver figura 6].30 La activación de p53 causa cierta detención en la fase G1 del ciclo celular, permitiendo a las células reparar el daño del ADN antes de continuar hacia la fase S y la replicación del ADN. En otras células, la activación de p53 ocasiona (a través de mecanismos no bien establecidos) apoptosis, un programa de suicidio celular en células cuyo ADN tiene daño irreparable. No es sorprendente que las mutaciones de p53 sean comunes en los cánceres humanos y se demuestren en hasta el 50 por ciento de los casos.31 La mayoría de las mutaciones son sustituciones de aminoácidos dentro del dominio de unión al ADN de p53, causando su plegamiento erróneo y unión a proteínas de choque térmico. En consecuencia, la velocidad de recambio de proteína se prolonga en estas moléculas p53 con mutación. Esto explica la paradoja de que los niveles elevados de proteína p53 en muestras tumorales se toman con frecuencia como evidencia de una mutación en el gen p53.
Las constelaciones de mutaciones p53 en los tumores han proporcionado también una correlación importante con el mecanismo supuesto de carcinogénesis. Por ejemplo, se han encontrado mutaciones p53 especificas en cánceres de hígado en una población en una región de China expuesta a niveles altos del carcinógeno hepático derivado de hongos aflatoxina, y el panel de mutaciones observadas en el cáncer pulmonar correlaciona con los inducidos por los carcinógenos policíclicos en el tabaco. Por el contrario, con frecuencia se detectan mutaciones p53 en el carcinoma de colon y recto en nucleótidos que son susceptibles a metilación, lo que parece ser un mecanismo común para las mutaciones espontáneas.32
La deleción de p53 en el ratón tiene efecto mínimo en el desarrollo normal, pero causa mucho mayor formación tumoral.33 Este cambio es más sutil en el ratón que no tiene un alelo p53, los ratones que carecen de ambos alelos sucumben rápido por tumores. La expresión exagerada de algunos mutantes p53 puede alterar la formación normal de tetrámeros p53, un fenotipo denominado dominante negativo o disfuncional, cauando un nivel intermedio de formación de tumores. En los humanos la pérdida de un alelo p53 de línea germinal es responsable del fenotipo multicáncer conocido como síndrome de Li-Fraumeni.34 Las familias afectadas por este raro rasgo autosómico dominante demostraron predisposición muy penetrante a diferentes tipos de cáncer, incluyendo sarcomas, cáncer de mama, tumores cerebrales y leucemia. El mecanismo por el que la incativación de la función de p53 produce desarrollo de cáncer parece ser la pérdida de un punto de revisión de la integridad del material genético antes de la replicación del ADN. p53 tiene también un papel crucial en desencadenar el suicidio celular en respuesta a señales de crecimiento inapropiadas, como las inducidas por la pérdida de otros supresores tumorales o por la activación de proto-oncogenes. En algunas neoplasias, como las que se originan en el síndrome de Li-Fraumeni, la pérdida de p53 inicia la tumorigénesis; en otras, como el cáncer de colon y recto, la inactivación de p53 es un evento genético tardío importante en la progresión del fenotipo de malignidad.
Como RB1, p53 es blanco específico de productos de oncogenes virales, incluyendo la proteína E1B del adenovirus, el antígeno T de SV40 y la proteína E6 del papilomavirus. Por lo tanto, los virus ADN tumorales combinan una estrategia dual: inducen proliferación celular al inactivar RB! y alteran la capacidad de p53 de desencadenar la muerte celular en respuesta a esta señal aberrante de proliferación.35 Se ha encontrado que algunas proteínas celulares controlan la función de p53. Entre estas se encuentran MDM2, una proteína que es inducida por p53 y que aumenta a su vez la degradación de p53, proporcionando un mecanismo crucial de retroalimentación negativa [ver figura 6]. El hecho de que p53 y MDM2 sean parte de una vía celular común es apoyado por alteraciones mutuamente excluyentes en los cánceres humanos: los osteosarcomas son causados por una mutación que inactiva p53 o amplifica el gen MDM2, provocando sobreproducción del producto del gen.36 Los ratones que carecen de ambos alelos de MDM2 mueren en etapa embrionaria temprana, a menos que también les falte p53, y en ese caso se desarrollan de modo normal hasta la edad adulta, lo que es evidencia de que MDM2 es indispensable para prevenir la actividad opuesta de p53 durante el desarrollo normal.37 Otro regulador potencialmente importante de p53 es el gen p19-ARF. Este gen no es habitual porque se sobrepone con el gen p16/INK4a pero es codificado por un marco de lectura de codón diferente.38 p19/ARF interactúa con MDM2 para regular el recambio de p53, su papel en el cáncer humano es sugerido por la frecuencia con la que estos tumores contienen deleciones genómicas que inactivan tanto p16/INK4a y p19-ARF. Por último, dos miembros de la familia genética de p53 (uno llamado p73 y el otro p63 o p51) tienen algunas de las mismas funciones de p53 in vitro, y su posible papel en el cáncer humano es motivo de investigación.39,40
Aunque p53 ha sido el gen supresor de tumores más estudiado de los implicados en la estabilidad genómica, otros genes asociados con enfermedades parecen funcionar en forma semejante o paralela. El gen de la ataxia-telangiectasia ATM, responsable de un rasgo autosómico recesivo caracterizado por degeneración cerebelosa, disfunción inmunológica y predisposición al cáncer, funciona hacia arriba de p53 y es necesario para su activación después de la radiación ionizante.41,42 Otros dos genes que pueden funcionar en la estabilidad genómica son el BRCA1 y el BRCA2.43,44 Los productos de estos genes, responsables de la predisposición al cáncer de mama y ovario, parecen interactuar con otras proteínas activas en la recombinación y reparación cromosómica.45 Aunque las propiedades funcionales precisas de los productos de los genes BRCA1 y BRCA2 se desconocen todavía, las mutaciones en estos genes pueden desencadenar transformación maligna al evitar que las células reparen el daño al ADN.
SUPRESORES DE TUMORES QUE PARTICIPAN EN LA SEÑALIZACION Y DIFERENCIACION
La identificación de genes supresores de tumores implicados en los síndromes con predisposición a cáncer ha llevado al descubrimiento de componentes clave en las vías de diferenciación celular. El gen WT1 codifica un regulador de la transcripción que se expresa en forma específica en los podocitos del glomérulo en desarrollo. Las mutaciones en el WT1 causan el tumor de Wilms, un cáncer renal embrionario y las mutaciones de línea germinal provocan defectos del desarrollo genitourinario. Los genes que normalmente son controlados por WT1 parecen participar en la proliferación y diferenciación.46 El gen von Hippel-Lindau (VHL) con frecuencia está mutado en cánceres de células renales en adultos y en la línea germinal de pacientes con un síndrome que incluye tumores vasculares tanto benignos como malignos. La proteína VHL parece funcionar en la regulación de la estabilidad de los ARN mensajeros específicos (ARNm), incluyendo el factor de crecimiento vascular epidérmico.47 El gen APC es un blanco clave en el cáncer de colon y recto: las mutaciones de línea germinal provocan poliposis colónica familiar, un síndrome caracterizado por el desarrollo de numerosos pólipos colónicos con un riesgo muy alto de transformación maligna. Las mutaciones somáticas constituyen el paso inicial en el desarrollo del cáncer de colon y recto.48 El producto APC normalmente se une e inactiva la beta-catenina, un cofactor importante en la señalización por la familia del gen WNT, que participa en vías de desarrollo claves. La inactivación del APC altera la función de WNT, causando señales de desarrollo anormal en el epitelio del colon. Los genes SMAD, activos en la señalización por el factor transformador de crecimiento-beta (FTC-beta) sufren mutación en los tumores pancreáticos.49 PTCH, un receptor para otro factor de crecimiento denominado puercoespín, sufre mutación en el síndrome de nevos de células basales, lo que causa predisposición a sufrir cáncer de piel.50 Otros genes supresores de tumores asociados con las vías de señalización celular son el gen NF1, que es responsable de la neurofibromatosis (enfermedad de von Recklinhausen) y cuyo producto genético normalmente inhibe al proto-oncogén ras,51 el gen NF2, que con frecuencia sufre mutación en los mesoteliomas y schwannomas y que codifica una proteína estructural que puede participar en la adhesión y proliferación celular,52 y el gen PTEN, que está implicado en un gran número de cánceres diferentes y en el síndrome con predisposición a cáncer de mama conocido como enfermedad de Cowden y que codifica una fosfatasa que se piensa cataliza la eliminación de una molécula de fosfato clave de proteínas blanco específicas.53 En resumen, los genes cuya alteración causan cáncer humano constituyen enlaces cruciales en las vías de diferenciación y proliferación celular.
Inestabilidad de microsatélites y reparación de errores en el ADN
El uso de tiras cortas y repetitivas de nucléosidos (microsatélites), que son muy variables de persona a persona, para rastrear diferentes alelos dentro de muestras de tumor ha permitido un gran descubrimiento: en algunos casos el tamaño de los microsatélites varía entre los tumores y las células normales del mismo pacientes. Esta observación sugiere que los tumores tienen genes inactivados que se requieren para corregir errores en la replicación de estas tiras cortas y repetitivas de ADN.54 La inactivación de los genes de reparación del ADN, que se conservan desde las bacterias a los humanos, provoca acúmulo de errores durante la replicación del ADN que pueden detectarse con marcadores de microsatélites colocados en forma aleatoria. Sin embargo, cuando ocurren errores en la replicación dentro de genes importantes el daño es grave y puede desencadenarse la proliferación maligna. Los miembros de la familia de genes de reparación de errores que son blanco más común de mutaciones en los cánceres humanos se denominan MSH2 y MLH1. Las mutaciones en estos genes causan pérdida de la función; sin embargo, los genes de reparación de errores difieren de los supresores de tumores clásicos porque su inactivación inicia el cáncer en forma indirecta (i.e., al aumentar la frecuencia de mutaciones de otros genes). La reintroducción de una copia de un gen de reparación normal después de que han surgido mutaciones secundarias parece demasiado tardía como para revertir el fenotipo maligno. Se han identificado los blancos potenciales de los genes de reparación de errores. Entre los más importantes están el receptor del FTC-beta, que contiene una secuencia corta y repetitiva dentro de su secuencia codificadora.55 Los tumores de colon y recto, en los que la mutación de un gen de reparación de errores caua inestabilidad de microsatélites, pueden tener mutaciones dentro de esta secuencia específica del gen del receptor del FTC-beta, alterando así una vía importante para la regulación de la proliferación celular en el epitelio del colon. Las mutaciones de línea germinal en los genes de reparación se asocian con un fenomeno de cáncer múltiple denominado síndrome de Lynch o cáncer de colon y recto hereditario no asociado a poliposis, que incluye predisposición a cáncer de colon, ovario y endometrio.56 Además, alrededor de 10% de los cánceres esporádicos de colon y recto tienen inestabilidad de microsatélites (denominada RER+), lo que sugiere que la alteración en la reparación de errores contribuye también al cáncer de colon y recto en ausencia de predisposición familiar [ver figura 7].57
Progresión tumoral
ACUMULO DE LESIONES GENETICAS
Puede requerirse una lesión genética en un solo gen para iniciar la transformación maligna. Sin embargo, la progresión del fenotipo maligno requiere de la adquisición de mutaciones adicionales que confieren una ventaja adicional de crecimiento y que, por lo tanto, son seleccionadas durante la expansión de la clona maligna [ver figura 8]. Un gen determinado puede ser determinante para la transformación en un solo tipo celular, pero tener un papel secundario en otro. El modelo más intensamente estudiado de progresión tumoral es el cáncer de colon y recto, para el que Kinzler y Vogelstein correlacionaron la progresión histológica de pólipo a carcinoma con el acúmulo de eventos genéticos.58 La mutación inactiva del gen APC se asocia con el desarrollo de hiperplasia epitelial, las mutaciones activadoras en el H-ras y los cambios en la metilación del ADN correlacionan con la progresión a pólipos adenomatosos, cambios en uno o más genes que residen en el cromosoma 18q indican la transición a adenomas de grado alto de malignidad y, por último, la inactivación de p53 acompaña la evolución a carcinoma maligno [ver figura 9]. Las lesiones preneoplásicas que causan el cáncer de colon y recto se definen con facilidad porque ocurren en la mucosa del colon y son accesibles a biopsia colonoscópica. Otros modelos de progresión tumoral son motivo de estudio, incluyendo los cánceres de esófago, cabeza y cuello, y vejiga.
MUERTE CELULAR E INMORTALIDAD
El estímulo de proliferación celular inicial que ocurre en un evento de transformación suele acompañarse por un aumento en la muerte celular o apoptosis, un mecanismo compensatorio que evita el crecimiento rápido de un cáncer. La inactivación del gen supresor de tumores p53 es la lesión genética más común que evita la respuesta de muerte celular, permitiendo que las células neoplásicas aumenten en número con rapidez. Otro gen que participa en la regulación de la apoptosis es bcl-2, que codifica una proteína mitocondrial que evita el inicio de la cascada de proteasas requerida para el suicidio celular.59 El papel de bcl-2 en el cáncer humano se ilustra mejor en el linfoma folicular de células B, un tumor indolente en el que una traslocación cromosómica hace que el gen bcl-2 quede bajo el control del gen promotor de inmunoglobulinas.60 La mayor expresión de bcl-2 en estas células linfoides impide su muerte celular programada, causando la mayor masa celular que caracteriza a este linfoma de crecimiento lento. Los modelos murinos en los que existe este rearreglo cromosómico tienen también un aumento marcado en el número de linfocitos, análogo al linfoma humano. bcl-2 es parte de una gran familia de genes, algunos miembros tienen efectos proapoptósicos y otros antagonizan la muerte celular programada.59 Un miembro proapoptósico de la familia bcl-2, BAX, contiene una secuencia repetitiva de nucleósidos que es blanco de mutaciones en tumores con inestabilidad de microsatélites.60,61 Por lo tanto, la expresión exagerada de los miembros de la familia bcl-2 antiapoptósicos o la inactivación de los genes proapoptósicos puede contribuir al fenotipo maligno.
Aunque la proliferación y la apoptosis constituyen respuestas celulares inmediatas y opuestas a las mutaciones de los proto-oncogenes y supresores de tumores, la inmortalidad célular se refiere a la vida celular ilimitada requerida para la formación de un tumor. Las células somáticas están programadas para sufrir solo un número limitado de divisiones celulares, después de lo cual entran en un estado latente. Esto es más aparente in vitro, en donde las células primarias cultivadas llegan al final de su vida programada, crecen y cesan de dividirse en forma permanente.62 Los mecanismos moleculares que causan este estado de latencia no se conocen bien, pero se ha sugerido que en modelos murinos los genes supresores de tumores p16-INK4a, p53 y p19-ARF pueden tener alguna función. Las células primarias que son estimuladas a proliferar por transformación oncógenica no tienen esta etapa latente y entran en crisis, o muerte celular masiva, al llegar al final de su vida. El pequeño número de células que sobrevive a la crisis es capaz de dividirse en forma infinita, constituyendo líneas celulares prácticamente inmortales. Estos estudios in vitro han proporcionado un modelo para estudiar el reloj biológico de las células somáticas normales, uno de los vigilantes más importantes contra la transformación maligna y que debe ser brincado por todas las células cancerosas destinadas a crecer más allá de su vida programada.
El reloj biológico celular se relaciona con los telómeros, los extremos de los cromosomas que se requieren para mantener la integridad cromosómica y que están compuestos de nucleótidos que se repiten.63 Debido a que la ADN polimerasa no copia los extremos finales de los cromosomas, un problema causado por el requerimiento de cebadores para iniciar la replicación de ADN, los telómeros se acortan con cada división celular. El periodo de latencia celular correlaciona bien con el acortamiento de los telómeros a menos del tamaño requerido para mantener la integridad de los cromosomas. En contraste con las células somáticas que tienen un periodo de vida finito, las células germinales y las células tronco hematopoyéticas conservan la longitud de sus telómeros con una enzima denominada telomerasa. La telomerasa es una ribonucleoproteína con actividad de transcriptasa inversa que usa el templete de ARN para agregar las repeticiones apropiadas de nucleótidos a los extremos de los telómeros, evitando así el acortamiento progresivo que acompaña a la división celular [ver figura 10].64,65 Los tumores humanos han adaptado el mecanismo fisiológico de evitar el periodo latente, y la mayoría de las células tumorales expresan niveles altos de telomerasa. Por analogía, las células cultivadas que surgen de la crisis y se han vuelto inmortales también expresan telomerasa, y esto se ha demostrado en modelos in vitro de transformación maligna. El aislamiento reciente de la subunidad catalítica de la telomerasa, denominada hTERT, ha permitido el análisis de su patrón de expresión a nivel unicelular, demostrando que comúnmente es inducida en las fases iniciales de la transformación maligna.66 El hecho de que hTERT codifica una transcriptasa inversa única ha estimulado el interés por diseñar agentes inhibitorios específicos.
METASTASIS Y ANGIOGENESIS
El acúmulo de mutaciones que causan proliferación celular y la adquisición de un periodo de vida infinito proporcionan los componentes esenciales de la transformación maligna, pero es la invasión tisular y la formación de metástasis lo que constituye las propiedades más visibles y agresivas del cáncer. Las células neoplásicas invaden los tejidos normales vecinos, penetrando la membrana basal epitelial y las células endoteliales y viajando a través de la circulación o el drenaje linfático hacia órganos distantes, en donde salen de los vasos y se establecen en sitios independientes para crecer. Por desgracia, los eventos genéticos responsables de estas complejas características del cáncer humano no se han definido y los modelos animales actuales han dado pocos datos hasta el momento. Se sabe que las células cancerígenas secretan enzimas, como la metaloproteinasa-2 de la matriz (MMP-2), que son capaces de digerir la membrana basal, facilitando la invasión tisular y la siembra en la circulación.67 La expresión de proteínas específicas de unión a la membrana, como la integrina alfavbeta3, parece anclar la MMP2 a la superficie de las células tumorales. En la actualidad se investiga en estudios clínicos la administración sistémica de inhibidores de la metaloproteinasa junto con fármacos quimioterápicos. También las moléculas de adhesión celular han sido implicadas en las metástasis, explicando en parte la predilección aparente de ciertos tumores por sitios de metástasis específicos. Entre las moléculas mejor caracterizadas está la proteína de superficie celular CD44; la ruptura alternativa del pre-ARNm produce variantes de transcritos de CD44, lo que produce diferentes isoformas de proteína. Existen variantes específicas de CD44 en niveles altos en las células tumorales metastásicas, que pueden tener un papel para facilitar la invasión.68 La menor expresión de E-cadherina y alfa5beta1 integrina en las células tumorales ha correlacionado también con mayor migración de las células y tumorigenicidad. Es posible que comparaciones adicionales entre tumores primarios y metastásicos revelen cambios genéticos que pudieran causar la progresión del fenotipo maligno.
Una propiedad crítica requerida para el crecimiento de los cánceres más allá de un tamaño mínimo es la formación de vasos sanguíneos.69 Los tumores varían en su grado de vascularización, e incluso dentro de un tumor determinado, ciertas áreas se vuelven hipóxicas y francamente necróticas al crecer en desproporción al aporte sanguíneo. Los péptidos angiogénicos secretados por los tumores son semejantes a los liberados durante la cicatrización fisiológica de las heridas, incluyendo el factor de crecimiento epidérmico vascular y el factor de crecimiento derivado de plaquetas. Sin embargo, en los modelos murinos ciertos tumores primarios pueden liberar también sustancias que se piensa evitan que sus metástasis a distancia recluten vasos sanguíneos, un fenómeno que podría explicarse como la prevención esencial de la competencia por nutrientes. Este descubrimiento llevó a Folkman y colaboradores a aislar dos péptidos angiogénicos denominados angiostatin y endostatin.70,71 Ambas sustancias son poco comunes porque parecen ser productos de degradación de otras proteínas, el plasminógeno y la colágena tipo XVIII. Su efecto en los modelos murinos es importante, causando necrosis de los tumores establecidos [ver figura 11]. Los tumores no desarrollan resistencia a estos agentes incluso después de su administración combinada y repetida, lo que sugiere que sus efectos son duraderos y persisten después de suspender el tratamiento.72 Los estudios en humanos de angiostatin y endostatina requieren de la síntesis de cantidades suficientes de estos novedosos agentes antiangiogénicos. La identificación de péptidos cortos que reconocen marcadores específicos dentro de las células endoteliales tumorales ha sugerido otro enfoque terapéutico novedoso que consiste en diseñar agentes quimioterápicos dirigidos contra el aporte vascular del tumor.73
RESISTENCIA A LOS FARMACOS QUIMIOTERAPICOS
La resección quirúrgica constituye la principal modalidad terapéutica para los tumores sólidos. La quimio y radioterapia son eficaces en el tratamiento de los tumores que se sabe tienen diseminación metastásica, como tratamiento adyuvante, en donde se sospecha depósito microscópico de tumor residual por el estado clínico del tumor y, en ocasiones, antes de la cirugía, cuando se requiere tratamiento sistémico para facilitar la resección de un tumor grande. Muchos agentes quimioterápicos derivan de compuestos que existen en la naturaleza, incluyendo agentes alquilantes que establecen enlaces químicos con el ADN, análogos de nucleótidos que inhiben las enzimas requeridas para la síntesis de ADN e inhibidores del haz mitótico. Los agentes terapéuticos que inducen daño directo al ADN típicamente se administran por infusión a corto plazo, mientras que los análogos de nucleótidos que se piensa son eficaces solo durante la fase S se administran por infusión continua para maximizar la exposición potencial de las células tumorales en reproducción. En general, los tumores con alta tasa de mitosis parecen tener mejor respuesta a la quimioterapia que los que tienen una fracción menor de células en proliferación. Los agentes quimioterápicos con frecuencia se administran en combinación, eligiéndose los fármacos con base en sus propiedades farmacológicas, el espectro de sensibilidad al fármaco mostrado por los diferentes tipos tumorales y la necesidad de evitar toxicidad similar para órganos como la médula ósea, el epitelio intestinal, el riñón y el sistema nervioso. Los intentos por probar los perfiles de sensibilidad a los fármacos de los tumores han sido limitados por el hecho de que solo puede cultivarse una pequeña fracción de las células de un tumor y es posible que éstas no representen en forma adecuada la heterogeneidad del tumor primario. La radioterapia consiste en administrar radiación ionizante, que causa rupturas en la doble cadena del ADN, a sitios anatómicos específicos infiltrados por el cáncer. La exposición de los tejidos normales se minimiza usando haces convergentes múltiples de radiación, junto con bloques protectores. En algunos cánceres, como el cáncer de recto localizado y el de ano, el uso combinado de quimio y radioterapia ha sido especialmente eficaz.
La resistencia del cáncer a los efectos de la quimioterapia puede ser intrínseca, como se observa con frecuencia en el melanoma, el cáncer hepatocelular y el carcinoma de células renales, que típicamente son refractarios a muchos agentes. En estos casos la resistencia a los fármacos puede reflejar las propiedades del tejido del que se originó el cáncer, como la necesidad de las células tubulares renales normales de protegerse de las toxinas que ocurren habitualmente y a las que se ven expuestas. En otros casos los cánceres que al inicio eran muy sensibles a la quimioterapia se vuelven resistentes, sea durante un curso de tratamiento o, con más frecuencia, en el momento de una recurrencia. Esta forma adquirida de resistencia a fármacos parece ser causada por mutaciones que confieren una ventaja selectiva al subgrupo de células tumorales, en forma análoga a otros eventos genéticos que contribuyen a la progresión tumoral. Usando modelos in vitro se han definido diversos mecanismos moleculares que confieren resistencia a agentes quimioterápicos específicos. Un ejemplo bien estudiado es el del metotrexate, que inhibe en forma específica la enzima dihidrofolato reductasa (DHFR) requerida para la síntesis de ADN. Los tumores pueden volverse resistentes al adquirir defectos en el transporte intracelular del metotrexate, por mutaciones en el gen DHFR, causando una enzima que no se une al metotrexate, o por amplificación del gen DHFR en sí, aumentando la cantidad de proteína elaborada, lo que supera la concentración intracelular del fármaco.74
Aunque los mecanismos de resistencia a fármacos específicos pueden contribuir a algunos casos de resistencia a la quimioterapia, la mayoría de las neoplasias parecen adquirir resistencia a una gama amplia de agentes, incluyendo algunos a los que no han tenido exposición previa. Este fenotipo de resistencia a múltiples fármacos puede reproducirse en cultivos celulares y llevó al descubrimiento de la familia de genes de resistencia a múltiples fármacos (mdr). Estos genes codifican bombas dependientes de adenosín trifosfato (ATP) que atraviesan la membrana celular y parecen participar en el transporte de compuestos orgánicos grandes.75 Otras miembros de esta familia de genes son el gen de la fibrosis quística y un gen procariote que puede contribuir a la resistencia a la quinina en el paludismo. El gen más estudiado implicado en la resistencia a la quimioterapia del cáncer es mdr-1, cuyo producto es capaz de exportar un amplio rango de compuestos orgánicos, incluyendo antraciclinas y alcaloides de la vinca. Los mecanismos moleculares que explican la especificidad al fármaco por los transportadores relacionados al mdr-1 se desconocen. Además, otros medicamentos, como ciclosporina y verapamil, son capaces de bloquear en forma no competitiva la extracción de los agentes quimioterapéuticos por el mdr-1 y se están incluyendo en esquemas para probar en estudios clínicos. La intensa relación entre la expresión exagerada de mdr-1 y la resistencia a la quimioterapia que es evidente in vitro no ha sido confirmada en la clínica, y la contribución de este gen a la mayoría de los casos de resistencia clínica a fármacos se desconoce aún. Sin embargo, en algunos cánceres, como el mieloma múltiple, la exposición repetida a antraciclinas y alcaloides de la vinca parece relacionarse con la mayor expresión de mdr-1 y resistencia a la quimioterapia,76 y la expresión mdr-1 parece ser un marcador de mal pronóstico en la leucemia mielógena aguda.77
El mayor conocimiento sobre la resistencia a agentes quimioterápicos proviene de la mejor comprensión del mecanismo de acción de estos agentes. El daño al ADN causado por los agentes quimioterápicos y la radiación ionizante ocasiona la activación de p53, causando muerte celular programada o apoptosis [ver figura 12]. El conocimiento de que la exposición a agentes quimioterápicos inicia un programa de suicidio celular activo, más que solo la muerte por daño masivo al ADN, aumenta la posibilidad de que estas vías apoptósicas estén alteradas en las células neoplásicas con resistencia a fármacos. En los modelos murinos las células estimuladas para proliferar por expresión de oncogenes sufren apoptosis después del tratamiento con quimioterápicos o radiación.77,78 Sin embargo, la inactivación de p53 en estas células se asocia con resistencia a estos agentes terapéuticos. La pérdida de p53 es una característica común de los cánceres humanos y diversos estudios han sugerido una correlación entre la inactivación de p53 y la resistencia a los agentes quimioterápicos, así como una evolución clínica más agresiva.32 En la actualidad se estudia la posibilidad de usar lesiones genéticas en los tumores para aumentar, en lugar de disminuir, la destrucción selectiva de células neoplásicas humanas. Un enfoque promisorio ha sido el desarrollo de un adenovirus modificado, capaz de replicarse e inducir lisis sólo en las células cuyos genes p53 hayan sido inactivados, destruyendo en forma específica las células neoplásicas [ver figura 13].79
Implicaciones clínicas del análisis genético
El análisis genético molecular del cáncer humano ha puesto al descubierto los mecanismos que explican la transformación maligna y la progresión desde lesiones premalignas tempranas hasta carcinomas invasores. La velocidad de los descubrimientos probablemente se acelerarán debido a las novedosas técnicas que facilitan la identificación de diferencias genéticas entre las células normales y las neoplásicas. El reto consistirá en aplicar esta información molecular al manejo clínico del cáncer humano.80 Aunque este campo aún inicia, diversos efoques son motivo de investigación activa. El diagnóstico temprano del cáncer actualmente se basa en vigilancia radiográfica, como en el caso de la mamografía y radiografía de tórax, la prueba de marcadores, como el antígeno prostático específico y el CA125 y procedimientos invasivos como cistoscopía y colonoscopía. Los marcadores moleculares en células que son desprendidas en el esputo, orina o heces podrán eventualmente convertirse en métodos de escrutinio exactos y eficaces.81-83 El uso apropiado de análisis de mutación de línea germinal permitirá también identificar a las personas con mayor riesgo de cáncer específicos que puedan beneficiarse con una vigilancia más agresiva.84 Las lesiones genéticas en las células malignas se han empleado también para facilitar el diagnóstico en casos en los que el análisis histológico no es definitivo y para detectar enfermedad residual mínima que no es apreciable por métodos estándar.85,86 El análisis de tumores en busca de lesiones genéticas que implican pronóstico se ha vuelto una conducta habitual en muchas leucemias.12-14 Los marcadores moleculares asociados con la respuesta a la quimioterapia, lo mismo que con probabilidad de diseminación metastásica, podrán guiar eventualmente el tratamiento de los tumores sólidos. Por último, la comprensión de las vías moleculares aberrantes que causan proliferación celular, reclutamiento de vasos y diseminación metastásica en los diferentes tipos de cáncer eventualmente permitirán el desarrollo de esquemas de tratamiento más específicos y eficaces.
Reconocimientos
Figuras 1 a 7 y 9 a 12 Seward Hung.
Figura 8 Janet Betries.
Tablas 1 y 2 De "The Promise of Cancer Genetics," por D. Haber y E. Fearon, en Lancet 351 (suppl II):1, 1998. Usado con autorización.
DR. DANIEL A. HABER, Ph.D.
El cáncer como una enfermedad genética
La expansión clonal no controlada de una célula, que con frecuencia causa invasión de los tejidos circundantes y diseminación metastásica, produce el cáncer. Las alteraciones genéticas iniciales en esta célula que desencadenan la proliferación aberrante son seguidas por acúmulo de mutaciones adicionales entre su progenie. Por último, ocurre un proceso de selección por el que las subclonas con propiedades de mayor crecimiento se vuelven dominantes dentro del tumor, la llamada progresión tumoral. En algunos tumores se ha definido una evolución genética histológica y molecular clara desde las lesiones precancerosas hasta el cáncer francamente maligno e invasor (v.gr., cánceres de cólon y vejiga). Sin embargo, en muchas neoplasias este proceso puede no ser clínicamente evidente. Los genes que están alterados en el desarrollo y progresión del cáncer aún se investigan en forma intensa. En muchos casos la identificación de estos genes ha contribuido a un mayor conocimiento de los mecanismos fisiológicos normales que controlan la proliferación celular. Los productos de estos genes participan en el control del ciclo celular básico, la trasmisión de señales de crecimiento, la regulación de la diferenciación y muerte celular, y en el establecimiento de la inmortalidad celular. Típicamente ocurre alteración de estos genes en las células somáticas destinadas a ser cancerosas. Sin embargo, en raros casos las mutaciones pueden trasmitirse en línea germinal, causando una predisposición genética al cáncer (i.e., síndromes familiares de cáncer).
Se piensa que los factores ambientales también contribuyen en el desarrollo del cáncer. Las interacciones entre factores ambientales y variaciones genéticas sutiles que distinguen a los individuos pueden constituir un determinante importante en el riesgo de cáncer en la población general. En algunos casos existe una relación directa entre el efecto del carcinógeno sobre el ADN y mutaciones específicas en el gen supresor de tumores p53 (v.gr., exposición a tabaco o a la toxina de hongos aflatoxina). En otros casos, efectos más indirectos pueden aumentar el riesgo de desarrollar cáncer (v.gr., exposición a carcinógenos potentes como asbesto o exposiciones más sutiles, como tratamiento prolongado con estrógenos). La exposición a la radiación ionizante es otra causa de cáncer. La incidencia de leucemias y de varios tumores sólidos entre los sobrevivientes a la bomba atómica proporcionó datos para estudios clásicos sobre las dosis de radiación y sus consecuencias. Datos más recientes se han obtenido a partir de casos de complicaciones tardías de radioterapia o quimioteraapia radiomimética administradas a pacientes como tratamiento para una neoplasia inicial.
Por último, las infecciones virales se han relacionado con el desarrollo de cánceres específicos. Estos incluyen el carcinoma cervical causado por subtipos específicos de papilomavirus humano, el carcinoma hepatocelular resultado de la infección crónica con virus de la hepatitis B, el carcinoma nasofaríngeo y los linfomas en huéspedes inmunosuprimidos que se asocian con virus Epstein-Barr, la rara leucemia aguda de células T causada por el virus linfotrópico transformador de células T humanas de tipo I y, potencialmente, el sarcoma de Kaposi que se asocia con herpesvirus humano tipo 8. Con estas excepciones, la mayoría de los cánceres en humanos no son causados por una infección viral. Sin embargo, mucho de nuestro conocimiento sobre los genes humanos que participan en el cáncer derivó originalmente de estudios de virus que provocan tumores en pollos y roedores. La alteración por estos virus de genes celulares que participan en la proliferación celular llevó a la identificación de los oncogenes, proporcionando la primera clave para los eventos genéticos que causan el cáncer en los humanos.
Oncogenes y proto-oncogenes
MUTACIONES QUE CAUSAN ACTIVACION O MODIFICACION EN LA FUNCION
Los genes que causan cáncer, u oncogenes, fueron descubiertos al encontrar que genes específicos de retrovirus de pollos y roedores eran capaces de transformar las células normales de mamíferos en cultivos. Se demostró que estos genes virales transformadores eran homólogos de los genes de mamíferos (proto-oncogenes) que habían sido robados de la célula huésped durante la evolución viral por su capacidad para estimular la proliferación celular.1 Los cánceres humanos primarios, aunque no son causados por una infección viral, tienen alelos activados similares de proto-oncogenes. Entre los primeros oncogenes descubiertos fueron los que codifican proteínas que participan en forma directa en la trasmisión de las señales de proliferación celular. Estos incluyen receptores para factores de crecimiento que están activados en forma constitutiva, aunque responden a la presencia continua de un factor de crecimiento (v.gr., receptor del factor de crecimiento derivado de plaquetas o receptor del factor de crecimiento epidérmico) así como a moléculas que le inhiben (i.e., moléculas que actúan sobre una región del ADN que se localiza en el lado 3´de un gen) que normalmente cambian entre el estado activo e inactivo pero que están mutadas en las células neoplásicas para mantenerse en una posición permanente de activación (v.gr., H-ras, K-ras y N-ras). Los mecanismos por los que estas proto-oncogenes celulares normales se activan en los cánceres humanos incluyen mutaciones puntiformes, amplificación de genes y traslocación de cromosomas [ver tabla 1 y figura 1]. Debido a que causan propiedades funcionales nuevas o alteradas en la proteína que codifican y son genéticamente dominantes sobre el segundo alelo normal, estas mutaciones comúnmente son conocidas como de modificación de función.
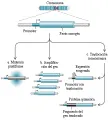
|
| Figura 1 |
| Mecanismo de activación de los proto-oncogenes |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
LLA-leucemia linfocítica aguda, LPA-leucemia promiellocítica aguda,LMC-leucemia mielógena crónica,EGF-factor de crecimiento epidérmico, FGF-factor de crecimiento de fibroblastos, GDNF,factor neurotrópico derivado de la línea de células gliales, GTPasa- guanosín trifosfatasa, HGF- factor de crecimiento de hepatocitos, CPCP-carcinoma celular de células pequeñas, ND- no determinado. |
Mutaciones puntiformes
Las mutaciones puntiformes que son capaces de activar un producto genético son poco comunes y reflejan propiedades funcionales específicas de la proteína en sí. Por ejemplo, solo las alteraciones específicas en los tres codones de la familia de proteínas ras causan una activación constitutiva de la señal ras.2 Se supone que otras mutaciones son funcionalmente silenciosas o causan proteínas inactivas y por lo tanto no son seleccionadas durante la transformación neoplásica. En forma semejante, cambios específicos en los aminoácidos de receptores de factores de crecimiento u otras moléculas de señal confieren un efecto activo o inductor de crecimiento o evitan la inhibición por parte de las señales fisiológicas apropiadas. De especial interés son las mutaciones en el gen ret, un receptor de factor de crecimiento y el único proto-oncogén encontrado que muta en la línea germinal de pacientes predispuestos a cáncer. Las sustituciones de aminoácidos con diferentes dominios funcionales de la proteína causan neoplasia endócrina múltiple tipo IIA o tipo IIB o cáncer medular de tiroides, mientras que las mutaciones inactivas provocan un defecto en el desarrollo que afecta la innervación del colon, la enfermedad de Hirschsprung.3 La estrecha relación entre las diferentes mutaciones en el gen ret y diversos síndromes neoplásicos y de desarrollo parece deberse a diversas propiedades funcionales que son mediadas por los varios dominios de la proteína ret así como por diferentes vías asociadas a ret que pueden activarse en distintos tipos celulares.
Amplificación de genes y traslocaciones cromosómicas
Además de las sustituciones de aminoácidos que causan un producto genético activado, los proto-oncogenes pueden convertirse a sus formas oncogénicas por alteraciones cromosómicas grandes. La amplificación genética, que consiste en la sobrerreplicación de fragmentos grandes de cromosomas, es un mecanismo común para la expresión exagerada aberrante de genes específicos en los tumores.4 Los genes amplificados, en ocasiones hasta en cientos de copias de los genes diploides normales, pueden residir en elementos extracromosómicos pequeños (cromosomas doble punto) o pueden integrarse dentro de una sola localización cromosómica, detectables como una región grande de tinción homogénea. En las células neoplásicas la pérdida de los genes celulares que contribuye normalmente al mantenimiento de la integridad genómica (v.gr., p53) puede facilitar los eventos cromosómicos aberrantes, incluyendo amplificación genética y, por lo tanto, acumulación de lesiones genéticas adicionales. Entre los oncogenes mejor conocidos que son amplificados por células neoplásicas está N-myc. Este factor de transcripción, que tiene un papel fisiológico en la estimulación de la proliferación celular, comunmente está amplificado en el neuroblastoma pediátrico, en donde confiere un mal pronóstico clínico.5 La expresión exagerada de los proto-oncogenes puede causar también una traslocación o rearreglo cromosómico, que mueve el gen de su promotor regulado fisiológicamente y lo coloca bajo el control de un promotor extraño y más activo.6 Por ejemplo, en el linfoma de Burkitt el locus cromosómico que contiene al gen c-myc se rearregla de modo que se pierden las regiones reguladoras colocadas en dirección superior (i.e., la región localizada del lado 5´del gen); la expresión del gen es dirigida por el intenso promotor de las cadenas pesadas de las inmunoglobulinas, que activa en forma constitutiva a los linfocitos B.7,8 La desregulación de la expresión de c-myc en estas células proporciona un potente estímulo para la proliferación celular.
La primera traslocación cromosómica específica identificada en el cáncer humano fue el cromosoma Filadelfia, que define a la leucemia mieloide crónica (LMC). La fusión de los cromosomas 9 y 22 causa la unión de dos genes no relacionados, el gen c-abl que codifica una tirosina cinasa, localizado en el cromosoma 9, y el gen bcr (por recombinación de punto de ruptura) localizado en el cromosoma 22.9 Por este rearreglo cromosómico se forma una proteína quimérica con nuevas propiedades de transformación. Las variantes poco comunes de LMC, en las que el cromosoma Filadelfia no es evidente por análisis citogenético, pueden tener rearreglos genéticos complejos, causando una traslocación bcr-abl que puede detectarse solo con marcadores moleculares. La fase acelerada o blástica de la LMC suele asociarse con duplicación del cromosoma Filadelfia, lo que sugiere que el aumento de copias de este gen aberrante confiere un efecto transformador dependiente de dosis. La fusión bcr-abl también demuestra una especificidad marcada al tipo celular: la clásica ruptura cromosómica se asocia con proliferación mieloide, mientras que una ruptura variante, que causa una alteración sutil en la proteína quimérica, origina la leucemia linfoide pediátrica, que tiene mal pronóstico.10 La importancia de la traslocación bcr-abl consiste en que el evento genético inicial en la LMC está apoyada por un modelo murino, en el que la expresión transgénica de la proteína quimérica bcr-abl en las células hematopoyéticas es suficiente para desencadenar proliferación tanto mieloide como linfoide.11
Otras traslocaciones cromosómicas que generan proteínas quiméricas nuevas se han asociado con diversos tipos de leucemia.12 En la leucemia promielocítica aguda (LPA), un rearreglo cromosómico une un gen nuevo, PML, con el gen del receptor-alfa del ácido retinoico (RAR-alfa). Aunque se desconocen las propiedades funcionales precisas de la molécula quimérica PML-RAR-alfa, esta traslocación puede explicar la respuesta dramática de esta leucemia al tratamiento con ácido retinoico-trans, que ha revolucionado el cuidado clínico de los pacientes con LPA.13 También se ha identificado otro producto de traslocación cromosómica nuevo, el TEL-AML1, que ocurre en una forma común y con buena respuesta a tratamiento de leucemia linfoblástica aguda pediátrica.14
La capacidad de las células leucémicas para crecer en cultivo lo suficiente para realizar un análisis citogenético ha facilitado la caracterización de las traslocaciones cromosómicas en las leucemias. Sin embargo, también se han observado traslocaciones cromosómicas específicas en tumores sólidos. Destacan el sarcoma de Ewing y la familia de tumores neuroepiteliales periféricos (TNEP). Estos tumores antes mal definidos se sabe que se asocian con una traslocación cromosómica que fusiona el gen EWS con diversos factores de transcripción de la familia del gen ETS (la proteína quimérica más común es EWS-FLI1).15 Este producto quimérico parece actuar directamente sobre los promotores para dirigir la expresión de genes que inducen la proliferación celular. La identificación de las traslocaciones EWS permitió la clasificación molecular de una clase de tumores cuya proliferación se debe a alteraciones genéticas semejantes y que responde a esquemas quimioterápicos parecidos.
FUNCION DE LOS ONCOGENES IN VITRO Y EN MODELOS MURINOS
La detección de mutaciones de cambio de función en los proto-oncogenes proporciona evidencia intensa de que estas alteraciones genéticas contribuyen a la transformación maligna. Sin embargo, el tiempo en que ocurren estas mutaciones durante el inicio y la progresión del cáncer puede ser variable. Con la sola excepción de c-ret, no se observan mutaciones de los proto-oncogenes en la línea germinal de los pacientes con síndromes familiares de cáncer, quizá porque tienen consecuencias adversas en el desarrollo normal. Las traslocaciones cromosómicas parecen constituir el evento inicial para la transformación maligna en algunas leucemias y tumores sólidos. En otros cánceres, las mutaciones en los proto-oncogenes pueden contribuir a la progresión del cáncer una vez que ya se ha desarrollado. Dos modelos experimentales, los ratones transgénicos y el cultivo celular, han sido de gran valor para reconocer las propiedades funcionales de los oncogenes. Los ratones transgénicos son generados por la inyección de plásmidos de ADN recombinante en un huevo fertilizado.16 Estos plásmidos pueden contener un oncogén cuya expresión esté dirigida por un promotor tisular específico. Una fracción de la progenie del ratón demostrará la expresión esperada del constructo transgénico. Aunque la expresión de ciertos oncogenes en los tejidos apropiados puede causar un crecimiento maligno, en otros casos el cáncer se desarrolla solo después del apareamiento de ratones que expresan diferentes oncogenes. La expresión de dos oncogenes diferentes en la progenie puede ayudar a inducir el crecimiento maligno.17 También se ha probado la función de oncogenes dominantes por medio de la transformación de células de roedores in vitro. Mientras que las líneas celulares establecidas requieren de poco estímulo para volverse tumorigénicas, las células cultivadas, como las derivadas de riñón de rata bebé o de fibroblastos de embrión de ratón, se transforman solo después de la introducción de combinaciones de potentes oncogenes.18 Por ejemplo, la combinación de c-myc y H-ras activado es eficaz para transformar a las células primarias, pero ninguno de estos dos oncogenes es suficiente en forma aislada.
Además de su papel para validar la posibilidad de transformación de los proto-oncogenes activados, los ensayos funcionales sobre el estudio de los virus ADN tumorales han proporcionado conocimiento importante sobre la relación entre los oncogenes y otra clase de genes relacionados al cáncer, los supresores de tumores. A diferencia de los retrovirus que tienen versiones activadas de proto-oncogenes celulares incorporadas a sus propios genomas, los virus ADN tumorales, como el adenovirus, el SV40 y el papilomavirus, codifican proteínas virales originales capaces de transformar a las células de roedores, monos o humanos. La introducción del antígeno SV40 T a ratones transgénicos causa el desarrollo de tumores en prácticamente cualquier tejido en el que se exprese. En forma semejante, la combinación de las proteínas adenovirales E1A y E1B es altamente transformadora en ensayos de trasnformación celular primaria. Un descubrimiento importante respecto a la etiología del cáncer fue el hallazgo de que los genes transformadores derivados de estos tres virus ADN tumorales no relacionados tenían la misma función, que consiste en la inactivación de los genes celulares supresores de tumores p53 y RB1.19 Por lo tanto, el cáncer se origina tanto por aumento de señales de proliferación como por pérdida de genes que inhiben la proliferación celular.
Genes supresores de tumores
REVERSION DE LA TUMORIGENICIDAD
Antes conocidos como anti-oncogenes, los genes supresores de tumores constituyen una familia de genes celulares cuya inactivación durante la tumorigénesis promueve el crecimiento maligno [ver tabla 2]. Su descubrimiento inicial surgió de tres líneas convergentes de investigación: el análisis de las propiedades funcionales de las células normales contra las cancerígenas, estudios epidemiológicos de cáncer en niños y el análisis genético molecular de pérdidas cromosómicas en tumores. Uno de los descubrimientos iniciales fue que la fusión de una célula maligna a una maligna causaba una célula híbrida que perdía sus propiedades malignas [ver figura 2].20 Este dato inesperado indicaba que el estado de malignidad era recesivo y sugería que genes en la célula no maligna podrían restablecer el control del crecimiento normal de una célula maligna que aparentemente había perdido información genética. Estos experimentos fueron seguidos por la demostración de que cromosomas individuales aislados de células normales eran capaces de revertir la tumorigenicidad de las células neoplásicas.21 La clonación molecular de genes específicos supresores de tumores hizo posible después demostrar que los plásmidos de ADN recombinante que contenían estos genes eran capaces de suprimir el crecimiento tumoral.
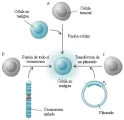
|
| Figura 2 |
| Reversión de la tumorigenicidad por genes supresores de tumores. |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
LMA-leucemia mieloide aguda, TGF-factor trasformador de crecimiento beta, WAGR-síndrome de tumor de Wilms, aniridia, alteraciones genitourinariasy retraso mental. |
EL MODELO DE KNUDSON Y LAS MUTACIONES DE PERDIDA DE FUNCION
La mayoría de los cánceres se originan durante la edad adulta; al parecer el aumento en la incidencia de cáncer al aumentar la edad es un reflejo del acúmulo del daño genético en las células tronco viejas. Los cánceres pediátricos, aunque raros, típicamente se desarrollan en células tronco jóvenes, como las células tronco renales que originan el tumor de Wilms, las células primitivas del neuroectodermo que se transforman en neuroblastoma y los retinoblastos que pueden producir el tumor ocular retinoblastoma. Un dato característico de estos tumores pediátricos es la existencia de familias en las que aproximadamente la mitad de los niños desarrollan tipos específicos de tumores. Dentro de estas familias con predisposición al cáncer los tumores que se originan con frecuencia son bilaterales o multicéntricos desde el principio y se desarrollan antes que lo correspondiente al comparar con los casos aislados en la población general. Al usar la distribución de Poisson para determinar la probabilidad de desarrollar cáncer como una función de la edad en los cánceres familiares comparado con los cánceres pediátricos esporádicos, Alfred Knudson propuso el modelo que forma ahora el fundamento de la genética del cáncer humano [ver figura 3].22
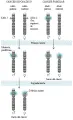
|
| Figura 3 |
| Modelo de Knudson de las dos lesiones para el inicio de los tumores. |
El modelo Knudson predice que los niños con tumores familiares han heredado una alteración genética inicial y requieren solo otra alteración genética adicional para iniciar la tumorigénesis. Por el contrario, los niños con tumores esporádicos necesitan adquirir dos alteraciones genéticas independientes dentro de la misma célula, un evento poco probable que explica porqué los cánceres esporádicos son menos frecuentes, de presentación unilateral y de inicio más tardío. Estudios genéticos subsecuentes en dos de los tumores estudiados por Knudson identificaron estos llamados golpes o alteraciones genéticas como la inactivación secuencial de los dos alelos de un gen supresor de tumores crítico: el RB1 en el retinoblastoma23 y el WT1 en el tumor de Wilms.24 El modelo Knudson explica también la paradoja respecto a que las mutaciones en genes supresores de tumores son de pérdida de función o recesivas [ver tabla 2], a pesar de que el cáncer familiar se presenta con un rasgo autosómico dominante. Aunque la pérdida de un solo alelo de un gen supresor de tumores puede ser funcionalmente silenciosa en presencia de un segundo alelo normal, la frecuencia de mutaciones espontáneas es suficientemente alta para asegurar que por lo menos una célula dentro de la célula blanco tenga posibilidad de perder un segundo alelo e iniciar la transformación maligna [ver figura 3].
Los genes supresores de tumores fueron identificados por la aplicación de herramientas citogenéticas y de genética molecular a estas observaciones funcionales y epidemiológicas. Mientras que la mutación inicial que inactiva un alelo de un gen supresor de tumor es típicamente una mutación puntiforme dentro de un gen en sí, la pérdida de un segundo alelo frecuentemente causa una deleción cromosómica o rearreglo mayor.25 La segunda alteración puede identificarse con facilidad por análisis de cariotipo, pero con más frecuencia se requiere análisis molecular para detectar la pérdida de marcadores polimórficos asociados en forma estrecha con el gen blanco. Los marcadores polimórficos (como los fragmentos de restricción de longitud polimórfica) son variables genéticas que pueden usarse para identificar cromosomas heredados de un padre [ver figura 4]. La pérdida de un polimorfismo derivado de cualquiera de los padres (llamada pérdida de la heterocigosidad [PH]) es indicación de que un gen supresor de tumores vecino ha sido inactivado en un cáncer. Estas pérdidas alélicas, que pueden mapearse en todos los cromosomas para un tipo determinado de tumor, han sido más eficaces para aislar genes supresores de tumores individuales, típicamente con estrategias de clonación posicional.

|
| Figura 4 |
| Pérdida de alelos en los tumores y pérdida de la heterorocigosidad (PH). |
PROGRESION DEL CICLO CELULAR Y VIA DEL RB1
El primer supresor de tumores identificado fue el gen RB1, que participa en el tumor ocular pediátrico retinoblastoma.23 De muchas maneras, el RB1 sigue siendo el prototipo de esta clase de genes y su conexión íntima con el mecanismo básico de progresión del ciclo celular ilustra la estrecha relación entre la proliferación celular normal y la transformación maligna. Como lo predice el modelo Knudson,22 un alelo de RB1 es inactivado en la línea germinal de niños con retinoblastoma familiar. Sus tumores demuestran PH, lo que indica pérdida somática del alelo restante. Los niños con retinoblastoma esporádico, que constituyen el 90 por ciento de los casos, tienen ambos alelos RB1 inactivados dentro de una sola célula somática. Los niños que portan una mutación en un alelo RB1 también tienen predisposición a osteosarcomas, aunque no tienen mayor susceptibilidad a otros tipos de cáncer en los que se ha demostrado pérdida somática del RB1 (v.gr., cáncer de pulmón). Esto ilustra una paradoja importante en la genética del cáncer: un gen expresado en todos los tejidos normales controla el inicio del cáncer solo en órganos seleccionados, en otros parece ser solo una más de muchas alteraciones genéticas que contribuyen al estado de neoplasia. Las explicaciones actuales para esta paradoja incluyen diferencias potenciales en el ensamble genético de diferentes tipos célulares, así como diferencias en los mecanismos compensatorios que pueden activarse después de la pérdida de un gen supresor de tumores (v.gr., el desencadenamiento de la muerte celular).
La proteína codificada por RB1 es una fosfoproteína nuclear que se une a productos de una familia de genes llamada E2F, que a su vez se asocian con proteínas de la familia DP.26 Los complejos E2F-DP tienen un papel crucial al activar la transcripción de los genes requeridos para la síntesis de ADN. La unión a RB1 suprime este efecto, esencialmente al bloquear a las células en la fase G1 del ciclo celular. Durante la proliferación celular normal, un complejo de cinasa que regula el ciclo celular y que contiene ciclina D y cinasa 4 dependiente de ciclina (DDK4) fosforila a RB1, causando su liberación del complejo E2F-DP, lo que permite que la célula entre a la fase de síntesis (S) del ADN del ciclo celular. Aunque la inactivación fisiológica de la RB1 a través de la hiperfosforilación es transitoria y reversible, la inactivación de RB1 por mutación, común en muchos tipos diferentes de tumores, permite un estímulo constante para la proliferación celular. En forma semejante, los virus ADN oncogénicos codifican proteínas que inactivan de modo específico e irreversible al producto del gen RB1, como la proteína E1A del adenovirus, el antígeno T del SV40 y la proteína E7 del papilomavirus. El RB1 en sí es un miembro de una familia de genes, con dos genes muy relacionados denominados p107 y p130. Aún no se sabe por qué el RB1 es un blanco frecuente de mutaciones en los cánceres humanos mientras que estos genes similares no mutan. El mejor conocimiento en las diferencias sutiles de la función de estos otros genes podrá explicar este hecho.
El estudio sobre la regulación normal del RB1 ha proporcionado también conocimientos importantes sobre otros genes relacionados con el cáncer que participan en la regulación del ciclo celular [ver figura 5]. El ciclin D, un gen cuya expresión causa hiperfosforilación e inactivación de RB1, suele amplificarse y expresarse en forma exagerada en el cáncer de mama. Por el contrario, p16/INK4a, un inhibidor de CDK4 que favorece la forma activa hiperfosforilada de RB1, sufre deleción en gran variedad de tumores. Las mutaciones de línea germinal de p16/INK4a son causa del melanoma familiar.22 Por último, también se han demostrado mutaciones en el gen cinasa CDK4 que hacen que la proteína codificada sea insensible a la inhibición por p16/INK4a, favoreciendo la inactivación de RB1, en cánceres humanos y en líneas de melanoma. Un dato característico de las mutaciones en estos diferentes componentes del ciclo celular es que parecen ser mutuamente excluyentes. Un solo tumor tiene una alteración en solo uno de estos genes, lo que es evidencia de que estas mutaciones son funcionalmente equivalentes y que no se confiere ninguna mayor ventaja de crecimiento por el acúmulo de mutaciones adicionales en componentes de la misma vía.28
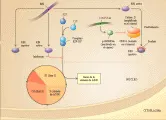
|
| Figura 5 |
| Gen del retinoblastoma (RB1) en el ciclo celular. |
GENES DE ESTABILIDAD GENOMICA: p53 y BRCA1
Aunque el gen supresor de tumores RB1 es un componente central de la regulación del ciclo celular, p53 tiene un papel crucial en el mantenimiento de la integridad genómica, de ahí su designación popular como el "guardián del genoma".29 p53 se expresa normalmente en niveles bajos en todas las células. Sin embargo, las lesiones genéticas, como la radiación ionizante, inician la estabilización y activación de la proteína p53. La p53 funciona como un factor de transcripción, dirigiendo la expresión de p21, un inhibidor de cinasas dependiente de ciclinas que regula el ciclo celular [ver figura 6].30 La activación de p53 causa cierta detención en la fase G1 del ciclo celular, permitiendo a las células reparar el daño del ADN antes de continuar hacia la fase S y la replicación del ADN. En otras células, la activación de p53 ocasiona (a través de mecanismos no bien establecidos) apoptosis, un programa de suicidio celular en células cuyo ADN tiene daño irreparable. No es sorprendente que las mutaciones de p53 sean comunes en los cánceres humanos y se demuestren en hasta el 50 por ciento de los casos.31 La mayoría de las mutaciones son sustituciones de aminoácidos dentro del dominio de unión al ADN de p53, causando su plegamiento erróneo y unión a proteínas de choque térmico. En consecuencia, la velocidad de recambio de proteína se prolonga en estas moléculas p53 con mutación. Esto explica la paradoja de que los niveles elevados de proteína p53 en muestras tumorales se toman con frecuencia como evidencia de una mutación en el gen p53.
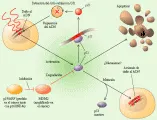
|
| Figura 6 |
| Vía de respuesta del p53 al daño del ADN. |
Las constelaciones de mutaciones p53 en los tumores han proporcionado también una correlación importante con el mecanismo supuesto de carcinogénesis. Por ejemplo, se han encontrado mutaciones p53 especificas en cánceres de hígado en una población en una región de China expuesta a niveles altos del carcinógeno hepático derivado de hongos aflatoxina, y el panel de mutaciones observadas en el cáncer pulmonar correlaciona con los inducidos por los carcinógenos policíclicos en el tabaco. Por el contrario, con frecuencia se detectan mutaciones p53 en el carcinoma de colon y recto en nucleótidos que son susceptibles a metilación, lo que parece ser un mecanismo común para las mutaciones espontáneas.32
La deleción de p53 en el ratón tiene efecto mínimo en el desarrollo normal, pero causa mucho mayor formación tumoral.33 Este cambio es más sutil en el ratón que no tiene un alelo p53, los ratones que carecen de ambos alelos sucumben rápido por tumores. La expresión exagerada de algunos mutantes p53 puede alterar la formación normal de tetrámeros p53, un fenotipo denominado dominante negativo o disfuncional, cauando un nivel intermedio de formación de tumores. En los humanos la pérdida de un alelo p53 de línea germinal es responsable del fenotipo multicáncer conocido como síndrome de Li-Fraumeni.34 Las familias afectadas por este raro rasgo autosómico dominante demostraron predisposición muy penetrante a diferentes tipos de cáncer, incluyendo sarcomas, cáncer de mama, tumores cerebrales y leucemia. El mecanismo por el que la incativación de la función de p53 produce desarrollo de cáncer parece ser la pérdida de un punto de revisión de la integridad del material genético antes de la replicación del ADN. p53 tiene también un papel crucial en desencadenar el suicidio celular en respuesta a señales de crecimiento inapropiadas, como las inducidas por la pérdida de otros supresores tumorales o por la activación de proto-oncogenes. En algunas neoplasias, como las que se originan en el síndrome de Li-Fraumeni, la pérdida de p53 inicia la tumorigénesis; en otras, como el cáncer de colon y recto, la inactivación de p53 es un evento genético tardío importante en la progresión del fenotipo de malignidad.
Como RB1, p53 es blanco específico de productos de oncogenes virales, incluyendo la proteína E1B del adenovirus, el antígeno T de SV40 y la proteína E6 del papilomavirus. Por lo tanto, los virus ADN tumorales combinan una estrategia dual: inducen proliferación celular al inactivar RB! y alteran la capacidad de p53 de desencadenar la muerte celular en respuesta a esta señal aberrante de proliferación.35 Se ha encontrado que algunas proteínas celulares controlan la función de p53. Entre estas se encuentran MDM2, una proteína que es inducida por p53 y que aumenta a su vez la degradación de p53, proporcionando un mecanismo crucial de retroalimentación negativa [ver figura 6]. El hecho de que p53 y MDM2 sean parte de una vía celular común es apoyado por alteraciones mutuamente excluyentes en los cánceres humanos: los osteosarcomas son causados por una mutación que inactiva p53 o amplifica el gen MDM2, provocando sobreproducción del producto del gen.36 Los ratones que carecen de ambos alelos de MDM2 mueren en etapa embrionaria temprana, a menos que también les falte p53, y en ese caso se desarrollan de modo normal hasta la edad adulta, lo que es evidencia de que MDM2 es indispensable para prevenir la actividad opuesta de p53 durante el desarrollo normal.37 Otro regulador potencialmente importante de p53 es el gen p19-ARF. Este gen no es habitual porque se sobrepone con el gen p16/INK4a pero es codificado por un marco de lectura de codón diferente.38 p19/ARF interactúa con MDM2 para regular el recambio de p53, su papel en el cáncer humano es sugerido por la frecuencia con la que estos tumores contienen deleciones genómicas que inactivan tanto p16/INK4a y p19-ARF. Por último, dos miembros de la familia genética de p53 (uno llamado p73 y el otro p63 o p51) tienen algunas de las mismas funciones de p53 in vitro, y su posible papel en el cáncer humano es motivo de investigación.39,40
Aunque p53 ha sido el gen supresor de tumores más estudiado de los implicados en la estabilidad genómica, otros genes asociados con enfermedades parecen funcionar en forma semejante o paralela. El gen de la ataxia-telangiectasia ATM, responsable de un rasgo autosómico recesivo caracterizado por degeneración cerebelosa, disfunción inmunológica y predisposición al cáncer, funciona hacia arriba de p53 y es necesario para su activación después de la radiación ionizante.41,42 Otros dos genes que pueden funcionar en la estabilidad genómica son el BRCA1 y el BRCA2.43,44 Los productos de estos genes, responsables de la predisposición al cáncer de mama y ovario, parecen interactuar con otras proteínas activas en la recombinación y reparación cromosómica.45 Aunque las propiedades funcionales precisas de los productos de los genes BRCA1 y BRCA2 se desconocen todavía, las mutaciones en estos genes pueden desencadenar transformación maligna al evitar que las células reparen el daño al ADN.
SUPRESORES DE TUMORES QUE PARTICIPAN EN LA SEÑALIZACION Y DIFERENCIACION
La identificación de genes supresores de tumores implicados en los síndromes con predisposición a cáncer ha llevado al descubrimiento de componentes clave en las vías de diferenciación celular. El gen WT1 codifica un regulador de la transcripción que se expresa en forma específica en los podocitos del glomérulo en desarrollo. Las mutaciones en el WT1 causan el tumor de Wilms, un cáncer renal embrionario y las mutaciones de línea germinal provocan defectos del desarrollo genitourinario. Los genes que normalmente son controlados por WT1 parecen participar en la proliferación y diferenciación.46 El gen von Hippel-Lindau (VHL) con frecuencia está mutado en cánceres de células renales en adultos y en la línea germinal de pacientes con un síndrome que incluye tumores vasculares tanto benignos como malignos. La proteína VHL parece funcionar en la regulación de la estabilidad de los ARN mensajeros específicos (ARNm), incluyendo el factor de crecimiento vascular epidérmico.47 El gen APC es un blanco clave en el cáncer de colon y recto: las mutaciones de línea germinal provocan poliposis colónica familiar, un síndrome caracterizado por el desarrollo de numerosos pólipos colónicos con un riesgo muy alto de transformación maligna. Las mutaciones somáticas constituyen el paso inicial en el desarrollo del cáncer de colon y recto.48 El producto APC normalmente se une e inactiva la beta-catenina, un cofactor importante en la señalización por la familia del gen WNT, que participa en vías de desarrollo claves. La inactivación del APC altera la función de WNT, causando señales de desarrollo anormal en el epitelio del colon. Los genes SMAD, activos en la señalización por el factor transformador de crecimiento-beta (FTC-beta) sufren mutación en los tumores pancreáticos.49 PTCH, un receptor para otro factor de crecimiento denominado puercoespín, sufre mutación en el síndrome de nevos de células basales, lo que causa predisposición a sufrir cáncer de piel.50 Otros genes supresores de tumores asociados con las vías de señalización celular son el gen NF1, que es responsable de la neurofibromatosis (enfermedad de von Recklinhausen) y cuyo producto genético normalmente inhibe al proto-oncogén ras,51 el gen NF2, que con frecuencia sufre mutación en los mesoteliomas y schwannomas y que codifica una proteína estructural que puede participar en la adhesión y proliferación celular,52 y el gen PTEN, que está implicado en un gran número de cánceres diferentes y en el síndrome con predisposición a cáncer de mama conocido como enfermedad de Cowden y que codifica una fosfatasa que se piensa cataliza la eliminación de una molécula de fosfato clave de proteínas blanco específicas.53 En resumen, los genes cuya alteración causan cáncer humano constituyen enlaces cruciales en las vías de diferenciación y proliferación celular.
Inestabilidad de microsatélites y reparación de errores en el ADN
El uso de tiras cortas y repetitivas de nucléosidos (microsatélites), que son muy variables de persona a persona, para rastrear diferentes alelos dentro de muestras de tumor ha permitido un gran descubrimiento: en algunos casos el tamaño de los microsatélites varía entre los tumores y las células normales del mismo pacientes. Esta observación sugiere que los tumores tienen genes inactivados que se requieren para corregir errores en la replicación de estas tiras cortas y repetitivas de ADN.54 La inactivación de los genes de reparación del ADN, que se conservan desde las bacterias a los humanos, provoca acúmulo de errores durante la replicación del ADN que pueden detectarse con marcadores de microsatélites colocados en forma aleatoria. Sin embargo, cuando ocurren errores en la replicación dentro de genes importantes el daño es grave y puede desencadenarse la proliferación maligna. Los miembros de la familia de genes de reparación de errores que son blanco más común de mutaciones en los cánceres humanos se denominan MSH2 y MLH1. Las mutaciones en estos genes causan pérdida de la función; sin embargo, los genes de reparación de errores difieren de los supresores de tumores clásicos porque su inactivación inicia el cáncer en forma indirecta (i.e., al aumentar la frecuencia de mutaciones de otros genes). La reintroducción de una copia de un gen de reparación normal después de que han surgido mutaciones secundarias parece demasiado tardía como para revertir el fenotipo maligno. Se han identificado los blancos potenciales de los genes de reparación de errores. Entre los más importantes están el receptor del FTC-beta, que contiene una secuencia corta y repetitiva dentro de su secuencia codificadora.55 Los tumores de colon y recto, en los que la mutación de un gen de reparación de errores caua inestabilidad de microsatélites, pueden tener mutaciones dentro de esta secuencia específica del gen del receptor del FTC-beta, alterando así una vía importante para la regulación de la proliferación celular en el epitelio del colon. Las mutaciones de línea germinal en los genes de reparación se asocian con un fenomeno de cáncer múltiple denominado síndrome de Lynch o cáncer de colon y recto hereditario no asociado a poliposis, que incluye predisposición a cáncer de colon, ovario y endometrio.56 Además, alrededor de 10% de los cánceres esporádicos de colon y recto tienen inestabilidad de microsatélites (denominada RER+), lo que sugiere que la alteración en la reparación de errores contribuye también al cáncer de colon y recto en ausencia de predisposición familiar [ver figura 7].57
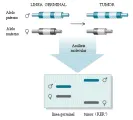
|
| Figura 7 |
| Inestabilidad de microsatélites y reparación del daño al ADN. |
Progresión tumoral
ACUMULO DE LESIONES GENETICAS
Puede requerirse una lesión genética en un solo gen para iniciar la transformación maligna. Sin embargo, la progresión del fenotipo maligno requiere de la adquisición de mutaciones adicionales que confieren una ventaja adicional de crecimiento y que, por lo tanto, son seleccionadas durante la expansión de la clona maligna [ver figura 8]. Un gen determinado puede ser determinante para la transformación en un solo tipo celular, pero tener un papel secundario en otro. El modelo más intensamente estudiado de progresión tumoral es el cáncer de colon y recto, para el que Kinzler y Vogelstein correlacionaron la progresión histológica de pólipo a carcinoma con el acúmulo de eventos genéticos.58 La mutación inactiva del gen APC se asocia con el desarrollo de hiperplasia epitelial, las mutaciones activadoras en el H-ras y los cambios en la metilación del ADN correlacionan con la progresión a pólipos adenomatosos, cambios en uno o más genes que residen en el cromosoma 18q indican la transición a adenomas de grado alto de malignidad y, por último, la inactivación de p53 acompaña la evolución a carcinoma maligno [ver figura 9]. Las lesiones preneoplásicas que causan el cáncer de colon y recto se definen con facilidad porque ocurren en la mucosa del colon y son accesibles a biopsia colonoscópica. Otros modelos de progresión tumoral son motivo de estudio, incluyendo los cánceres de esófago, cabeza y cuello, y vejiga.
|
|
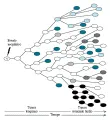

El estímulo de proliferación celular inicial que ocurre en un evento de transformación suele acompañarse por un aumento en la muerte celular o apoptosis, un mecanismo compensatorio que evita el crecimiento rápido de un cáncer. La inactivación del gen supresor de tumores p53 es la lesión genética más común que evita la respuesta de muerte celular, permitiendo que las células neoplásicas aumenten en número con rapidez. Otro gen que participa en la regulación de la apoptosis es bcl-2, que codifica una proteína mitocondrial que evita el inicio de la cascada de proteasas requerida para el suicidio celular.59 El papel de bcl-2 en el cáncer humano se ilustra mejor en el linfoma folicular de células B, un tumor indolente en el que una traslocación cromosómica hace que el gen bcl-2 quede bajo el control del gen promotor de inmunoglobulinas.60 La mayor expresión de bcl-2 en estas células linfoides impide su muerte celular programada, causando la mayor masa celular que caracteriza a este linfoma de crecimiento lento. Los modelos murinos en los que existe este rearreglo cromosómico tienen también un aumento marcado en el número de linfocitos, análogo al linfoma humano. bcl-2 es parte de una gran familia de genes, algunos miembros tienen efectos proapoptósicos y otros antagonizan la muerte celular programada.59 Un miembro proapoptósico de la familia bcl-2, BAX, contiene una secuencia repetitiva de nucleósidos que es blanco de mutaciones en tumores con inestabilidad de microsatélites.60,61 Por lo tanto, la expresión exagerada de los miembros de la familia bcl-2 antiapoptósicos o la inactivación de los genes proapoptósicos puede contribuir al fenotipo maligno.
Aunque la proliferación y la apoptosis constituyen respuestas celulares inmediatas y opuestas a las mutaciones de los proto-oncogenes y supresores de tumores, la inmortalidad célular se refiere a la vida celular ilimitada requerida para la formación de un tumor. Las células somáticas están programadas para sufrir solo un número limitado de divisiones celulares, después de lo cual entran en un estado latente. Esto es más aparente in vitro, en donde las células primarias cultivadas llegan al final de su vida programada, crecen y cesan de dividirse en forma permanente.62 Los mecanismos moleculares que causan este estado de latencia no se conocen bien, pero se ha sugerido que en modelos murinos los genes supresores de tumores p16-INK4a, p53 y p19-ARF pueden tener alguna función. Las células primarias que son estimuladas a proliferar por transformación oncógenica no tienen esta etapa latente y entran en crisis, o muerte celular masiva, al llegar al final de su vida. El pequeño número de células que sobrevive a la crisis es capaz de dividirse en forma infinita, constituyendo líneas celulares prácticamente inmortales. Estos estudios in vitro han proporcionado un modelo para estudiar el reloj biológico de las células somáticas normales, uno de los vigilantes más importantes contra la transformación maligna y que debe ser brincado por todas las células cancerosas destinadas a crecer más allá de su vida programada.
El reloj biológico celular se relaciona con los telómeros, los extremos de los cromosomas que se requieren para mantener la integridad cromosómica y que están compuestos de nucleótidos que se repiten.63 Debido a que la ADN polimerasa no copia los extremos finales de los cromosomas, un problema causado por el requerimiento de cebadores para iniciar la replicación de ADN, los telómeros se acortan con cada división celular. El periodo de latencia celular correlaciona bien con el acortamiento de los telómeros a menos del tamaño requerido para mantener la integridad de los cromosomas. En contraste con las células somáticas que tienen un periodo de vida finito, las células germinales y las células tronco hematopoyéticas conservan la longitud de sus telómeros con una enzima denominada telomerasa. La telomerasa es una ribonucleoproteína con actividad de transcriptasa inversa que usa el templete de ARN para agregar las repeticiones apropiadas de nucleótidos a los extremos de los telómeros, evitando así el acortamiento progresivo que acompaña a la división celular [ver figura 10].64,65 Los tumores humanos han adaptado el mecanismo fisiológico de evitar el periodo latente, y la mayoría de las células tumorales expresan niveles altos de telomerasa. Por analogía, las células cultivadas que surgen de la crisis y se han vuelto inmortales también expresan telomerasa, y esto se ha demostrado en modelos in vitro de transformación maligna. El aislamiento reciente de la subunidad catalítica de la telomerasa, denominada hTERT, ha permitido el análisis de su patrón de expresión a nivel unicelular, demostrando que comúnmente es inducida en las fases iniciales de la transformación maligna.66 El hecho de que hTERT codifica una transcriptasa inversa única ha estimulado el interés por diseñar agentes inhibitorios específicos.

|
| Figura 10 |
| Latencia celular y activación de la telomerasa. |
METASTASIS Y ANGIOGENESIS
El acúmulo de mutaciones que causan proliferación celular y la adquisición de un periodo de vida infinito proporcionan los componentes esenciales de la transformación maligna, pero es la invasión tisular y la formación de metástasis lo que constituye las propiedades más visibles y agresivas del cáncer. Las células neoplásicas invaden los tejidos normales vecinos, penetrando la membrana basal epitelial y las células endoteliales y viajando a través de la circulación o el drenaje linfático hacia órganos distantes, en donde salen de los vasos y se establecen en sitios independientes para crecer. Por desgracia, los eventos genéticos responsables de estas complejas características del cáncer humano no se han definido y los modelos animales actuales han dado pocos datos hasta el momento. Se sabe que las células cancerígenas secretan enzimas, como la metaloproteinasa-2 de la matriz (MMP-2), que son capaces de digerir la membrana basal, facilitando la invasión tisular y la siembra en la circulación.67 La expresión de proteínas específicas de unión a la membrana, como la integrina alfavbeta3, parece anclar la MMP2 a la superficie de las células tumorales. En la actualidad se investiga en estudios clínicos la administración sistémica de inhibidores de la metaloproteinasa junto con fármacos quimioterápicos. También las moléculas de adhesión celular han sido implicadas en las metástasis, explicando en parte la predilección aparente de ciertos tumores por sitios de metástasis específicos. Entre las moléculas mejor caracterizadas está la proteína de superficie celular CD44; la ruptura alternativa del pre-ARNm produce variantes de transcritos de CD44, lo que produce diferentes isoformas de proteína. Existen variantes específicas de CD44 en niveles altos en las células tumorales metastásicas, que pueden tener un papel para facilitar la invasión.68 La menor expresión de E-cadherina y alfa5beta1 integrina en las células tumorales ha correlacionado también con mayor migración de las células y tumorigenicidad. Es posible que comparaciones adicionales entre tumores primarios y metastásicos revelen cambios genéticos que pudieran causar la progresión del fenotipo maligno.
Una propiedad crítica requerida para el crecimiento de los cánceres más allá de un tamaño mínimo es la formación de vasos sanguíneos.69 Los tumores varían en su grado de vascularización, e incluso dentro de un tumor determinado, ciertas áreas se vuelven hipóxicas y francamente necróticas al crecer en desproporción al aporte sanguíneo. Los péptidos angiogénicos secretados por los tumores son semejantes a los liberados durante la cicatrización fisiológica de las heridas, incluyendo el factor de crecimiento epidérmico vascular y el factor de crecimiento derivado de plaquetas. Sin embargo, en los modelos murinos ciertos tumores primarios pueden liberar también sustancias que se piensa evitan que sus metástasis a distancia recluten vasos sanguíneos, un fenómeno que podría explicarse como la prevención esencial de la competencia por nutrientes. Este descubrimiento llevó a Folkman y colaboradores a aislar dos péptidos angiogénicos denominados angiostatin y endostatin.70,71 Ambas sustancias son poco comunes porque parecen ser productos de degradación de otras proteínas, el plasminógeno y la colágena tipo XVIII. Su efecto en los modelos murinos es importante, causando necrosis de los tumores establecidos [ver figura 11]. Los tumores no desarrollan resistencia a estos agentes incluso después de su administración combinada y repetida, lo que sugiere que sus efectos son duraderos y persisten después de suspender el tratamiento.72 Los estudios en humanos de angiostatin y endostatina requieren de la síntesis de cantidades suficientes de estos novedosos agentes antiangiogénicos. La identificación de péptidos cortos que reconocen marcadores específicos dentro de las células endoteliales tumorales ha sugerido otro enfoque terapéutico novedoso que consiste en diseñar agentes quimioterápicos dirigidos contra el aporte vascular del tumor.73
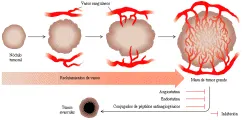
|
| Figura 11 |
| Angiogénesis tumoral y sus inhibidores. |
RESISTENCIA A LOS FARMACOS QUIMIOTERAPICOS
La resección quirúrgica constituye la principal modalidad terapéutica para los tumores sólidos. La quimio y radioterapia son eficaces en el tratamiento de los tumores que se sabe tienen diseminación metastásica, como tratamiento adyuvante, en donde se sospecha depósito microscópico de tumor residual por el estado clínico del tumor y, en ocasiones, antes de la cirugía, cuando se requiere tratamiento sistémico para facilitar la resección de un tumor grande. Muchos agentes quimioterápicos derivan de compuestos que existen en la naturaleza, incluyendo agentes alquilantes que establecen enlaces químicos con el ADN, análogos de nucleótidos que inhiben las enzimas requeridas para la síntesis de ADN e inhibidores del haz mitótico. Los agentes terapéuticos que inducen daño directo al ADN típicamente se administran por infusión a corto plazo, mientras que los análogos de nucleótidos que se piensa son eficaces solo durante la fase S se administran por infusión continua para maximizar la exposición potencial de las células tumorales en reproducción. En general, los tumores con alta tasa de mitosis parecen tener mejor respuesta a la quimioterapia que los que tienen una fracción menor de células en proliferación. Los agentes quimioterápicos con frecuencia se administran en combinación, eligiéndose los fármacos con base en sus propiedades farmacológicas, el espectro de sensibilidad al fármaco mostrado por los diferentes tipos tumorales y la necesidad de evitar toxicidad similar para órganos como la médula ósea, el epitelio intestinal, el riñón y el sistema nervioso. Los intentos por probar los perfiles de sensibilidad a los fármacos de los tumores han sido limitados por el hecho de que solo puede cultivarse una pequeña fracción de las células de un tumor y es posible que éstas no representen en forma adecuada la heterogeneidad del tumor primario. La radioterapia consiste en administrar radiación ionizante, que causa rupturas en la doble cadena del ADN, a sitios anatómicos específicos infiltrados por el cáncer. La exposición de los tejidos normales se minimiza usando haces convergentes múltiples de radiación, junto con bloques protectores. En algunos cánceres, como el cáncer de recto localizado y el de ano, el uso combinado de quimio y radioterapia ha sido especialmente eficaz.
La resistencia del cáncer a los efectos de la quimioterapia puede ser intrínseca, como se observa con frecuencia en el melanoma, el cáncer hepatocelular y el carcinoma de células renales, que típicamente son refractarios a muchos agentes. En estos casos la resistencia a los fármacos puede reflejar las propiedades del tejido del que se originó el cáncer, como la necesidad de las células tubulares renales normales de protegerse de las toxinas que ocurren habitualmente y a las que se ven expuestas. En otros casos los cánceres que al inicio eran muy sensibles a la quimioterapia se vuelven resistentes, sea durante un curso de tratamiento o, con más frecuencia, en el momento de una recurrencia. Esta forma adquirida de resistencia a fármacos parece ser causada por mutaciones que confieren una ventaja selectiva al subgrupo de células tumorales, en forma análoga a otros eventos genéticos que contribuyen a la progresión tumoral. Usando modelos in vitro se han definido diversos mecanismos moleculares que confieren resistencia a agentes quimioterápicos específicos. Un ejemplo bien estudiado es el del metotrexate, que inhibe en forma específica la enzima dihidrofolato reductasa (DHFR) requerida para la síntesis de ADN. Los tumores pueden volverse resistentes al adquirir defectos en el transporte intracelular del metotrexate, por mutaciones en el gen DHFR, causando una enzima que no se une al metotrexate, o por amplificación del gen DHFR en sí, aumentando la cantidad de proteína elaborada, lo que supera la concentración intracelular del fármaco.74
Aunque los mecanismos de resistencia a fármacos específicos pueden contribuir a algunos casos de resistencia a la quimioterapia, la mayoría de las neoplasias parecen adquirir resistencia a una gama amplia de agentes, incluyendo algunos a los que no han tenido exposición previa. Este fenotipo de resistencia a múltiples fármacos puede reproducirse en cultivos celulares y llevó al descubrimiento de la familia de genes de resistencia a múltiples fármacos (mdr). Estos genes codifican bombas dependientes de adenosín trifosfato (ATP) que atraviesan la membrana celular y parecen participar en el transporte de compuestos orgánicos grandes.75 Otras miembros de esta familia de genes son el gen de la fibrosis quística y un gen procariote que puede contribuir a la resistencia a la quinina en el paludismo. El gen más estudiado implicado en la resistencia a la quimioterapia del cáncer es mdr-1, cuyo producto es capaz de exportar un amplio rango de compuestos orgánicos, incluyendo antraciclinas y alcaloides de la vinca. Los mecanismos moleculares que explican la especificidad al fármaco por los transportadores relacionados al mdr-1 se desconocen. Además, otros medicamentos, como ciclosporina y verapamil, son capaces de bloquear en forma no competitiva la extracción de los agentes quimioterapéuticos por el mdr-1 y se están incluyendo en esquemas para probar en estudios clínicos. La intensa relación entre la expresión exagerada de mdr-1 y la resistencia a la quimioterapia que es evidente in vitro no ha sido confirmada en la clínica, y la contribución de este gen a la mayoría de los casos de resistencia clínica a fármacos se desconoce aún. Sin embargo, en algunos cánceres, como el mieloma múltiple, la exposición repetida a antraciclinas y alcaloides de la vinca parece relacionarse con la mayor expresión de mdr-1 y resistencia a la quimioterapia,76 y la expresión mdr-1 parece ser un marcador de mal pronóstico en la leucemia mielógena aguda.77
El mayor conocimiento sobre la resistencia a agentes quimioterápicos proviene de la mejor comprensión del mecanismo de acción de estos agentes. El daño al ADN causado por los agentes quimioterápicos y la radiación ionizante ocasiona la activación de p53, causando muerte celular programada o apoptosis [ver figura 12]. El conocimiento de que la exposición a agentes quimioterápicos inicia un programa de suicidio celular activo, más que solo la muerte por daño masivo al ADN, aumenta la posibilidad de que estas vías apoptósicas estén alteradas en las células neoplásicas con resistencia a fármacos. En los modelos murinos las células estimuladas para proliferar por expresión de oncogenes sufren apoptosis después del tratamiento con quimioterápicos o radiación.77,78 Sin embargo, la inactivación de p53 en estas células se asocia con resistencia a estos agentes terapéuticos. La pérdida de p53 es una característica común de los cánceres humanos y diversos estudios han sugerido una correlación entre la inactivación de p53 y la resistencia a los agentes quimioterápicos, así como una evolución clínica más agresiva.32 En la actualidad se estudia la posibilidad de usar lesiones genéticas en los tumores para aumentar, en lugar de disminuir, la destrucción selectiva de células neoplásicas humanas. Un enfoque promisorio ha sido el desarrollo de un adenovirus modificado, capaz de replicarse e inducir lisis sólo en las células cuyos genes p53 hayan sido inactivados, destruyendo en forma específica las células neoplásicas [ver figura 13].79
|
|
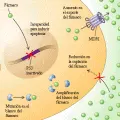
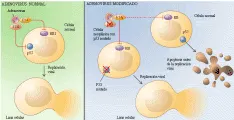
El análisis genético molecular del cáncer humano ha puesto al descubierto los mecanismos que explican la transformación maligna y la progresión desde lesiones premalignas tempranas hasta carcinomas invasores. La velocidad de los descubrimientos probablemente se acelerarán debido a las novedosas técnicas que facilitan la identificación de diferencias genéticas entre las células normales y las neoplásicas. El reto consistirá en aplicar esta información molecular al manejo clínico del cáncer humano.80 Aunque este campo aún inicia, diversos efoques son motivo de investigación activa. El diagnóstico temprano del cáncer actualmente se basa en vigilancia radiográfica, como en el caso de la mamografía y radiografía de tórax, la prueba de marcadores, como el antígeno prostático específico y el CA125 y procedimientos invasivos como cistoscopía y colonoscopía. Los marcadores moleculares en células que son desprendidas en el esputo, orina o heces podrán eventualmente convertirse en métodos de escrutinio exactos y eficaces.81-83 El uso apropiado de análisis de mutación de línea germinal permitirá también identificar a las personas con mayor riesgo de cáncer específicos que puedan beneficiarse con una vigilancia más agresiva.84 Las lesiones genéticas en las células malignas se han empleado también para facilitar el diagnóstico en casos en los que el análisis histológico no es definitivo y para detectar enfermedad residual mínima que no es apreciable por métodos estándar.85,86 El análisis de tumores en busca de lesiones genéticas que implican pronóstico se ha vuelto una conducta habitual en muchas leucemias.12-14 Los marcadores moleculares asociados con la respuesta a la quimioterapia, lo mismo que con probabilidad de diseminación metastásica, podrán guiar eventualmente el tratamiento de los tumores sólidos. Por último, la comprensión de las vías moleculares aberrantes que causan proliferación celular, reclutamiento de vasos y diseminación metastásica en los diferentes tipos de cáncer eventualmente permitirán el desarrollo de esquemas de tratamiento más específicos y eficaces.
Figuras 1 a 7 y 9 a 12 Seward Hung.
Figura 8 Janet Betries.
Tablas 1 y 2 De "The Promise of Cancer Genetics," por D. Haber y E. Fearon, en Lancet 351 (suppl II):1, 1998. Usado con autorización.
Bibliografía
- Stehelin D, Varmus HE, Bishop JM, et al: DNA related to transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 260:170, 1976 [PMID 176594]
- Lowy DR, Willumsen BM: Function and regulation of ras. Annu Rev Biochem 62:851, 1993 [PMID 8352603]
- Eng C, Clayton D, Schuffenecker I, et al: The relationship between specific net proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET Mutation Consortium analysis. JAMA 276:1575, 1996 [PMID 8918855]
- Schimke RT: Gene amplification, drug resistance, and cancer. Cancer Res 44:1735, 1984
- Brodeur GM, Seeger RC, Schwab M, et al: Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224:1121, 1984 [PMID 6719137]
- Rabbitts TH: Chromosomal translocations in human cancer. Nature 372:143, 1994
- Dalla-Favera R, Bregni M, Erikson J, et al: Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA 79:7824, 1982 [PMID 6961453]
- Taub R, Kirsch I, Morton C, et al: Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cell. Proc Natl Acad Sci USA 79:7837, 1982 [PMID 6818551]
- Shtivelman E, Lifshitz B, Gale RP, et al: Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 315:550, 1985 [PMID 2989692]
- Rowley JD: Biological implications of consistent chromosome rearrangements in leukemia and lymphoma. Cancer Res 44:3159, 1984
- Daley GQ, Van-Etten RA, Baltimore D: Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247:824, 1990 [PMID 2406902]
- Look AT: Oncogenic transcription factors in the human acute leukemias. Science 278:1059, 1997
- Warrell RP Jr, Frankel SR, Miller WH Jr, et al: Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med 324:1385, 1991 [PMID 1850498]
- Golub TR, Barker GF, Bohlander SK, et al: Fusion of the TEL gene on 12p13 to the AMLI gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA 92:4917, 1995 [PMID 7761424]
- Delattre O, Zucman J, Melot T, et al: The Ewing family of tumors: a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331:294, 1994 [PMID 8022439]
- Adams JM, Cory S: Transgenic models of tumor development. Science 254:1161, 1991 [PMID 1957168]
- Sinn E, Muller W, Pattengale PK, et al: Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 49:465, 1987 [PMID 3032456]
- Land H, Parada LF, Weinberg RA: Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304:596, 1983 [PMID 6308472]
- Whyte P, Buchkovich KJ, Horowitz JM, et al: Association between an oncogene and an antioncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 234:124, 1988
- Harris H, Klein G: Malignancy of somatic cell hybrids. Nature 224:1314, 1969
- Saxon PJ, Srivastan ES, Stanbridge EJ: Introduction of human chromosome 11 via microcell transfer controls tumorigenic expression of HeLa cells. EMBO J 5:3461, 1986 [PMID 2881780]
- Knudson AG Jr: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820, 1971
- Friend SH, Bernards R, Rogelij S, et al: A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323:643, 1986 [PMID 2877398]
- Call KM, Glaser T, Ito CY, et al: Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell 60:509, 1990 [PMID 2154335]
- Cavenee WK, Dryja TP, Phillips RA, et al: Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 305:779, 1983 [PMID 6633649]
- Weinberg RA: The Rb protein and cell cycle control. Cell 81:323, 1995
- Hussussian CJ, Struewing JP, Goldstein AM, et al: Germline p16 mutations in familial melanoma. Nat Genet 8:15, 1994 [PMID 7987387]
- Sherr CJ: Cancer cell cycles. Science 274:1672, 1996
- Lane DP: p53, guardian of the genome. Nature 358:15, 1992
- El-Deiry WS, Tokino T, Velculescu VE, et al: WAF1, a potential mediator of p53 tumor suppression. Cell 75:817, 1993 [PMID 8242752]
- Hollstein M, Sidransky D, Vogelstein B, et al: p53 mutations in human cancers. Science 253:49, 1991 [PMID 1905840]
- Harris CC, Hollstein M: Clinical implications of the p53 tumor suppressor gene. N Engl J Med 329:1318, 1993 [PMID 8413413]
- Donehower LA, Harvey M, Slagle BL, et al: Mice deficient of p53 are developmentally normal but susceptible to spontaneous tumors. Nature 356:215, 1992 [PMID 1552940]
- Malkin D, Li FP, Strong LC, et al: Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233, 1990 [PMID 1978757]
- Debbas M, White E: Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 7:546, 1993 [PMID 8384580]
- Oliner JD, Kinzler KW, Meltzer PS, et al: Amplifications of a gene encoding a p53-associated protein in human sarcoma. Nature 358:80, 1992 [PMID 1614537]
- Montes ede Oca Luna R, Wagner DS, Lozano G: Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378:203, 1995
- Quelle DE, Zindy F, Ashmun RA, et al: Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83:993, 1995 [PMID 8521522]
- Kaghad M, Bonnet H, Yang A, et al: Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90:809, 1997 [PMID 9288759]
- Osada M, Ohba M, Kawahara C, et al: Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med 4:839, 1998 [PMID 9662378]
- Kastan MB, Zhan Q, el-Deiry WS, et al: A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71:587, 1992 [PMID 1423616]
- Savitsky K, Bar-Shira A, Gilad S, et al: A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268:1749, 1996
- Miki Y, Swensew J, Shattuck-Eidens D, et al: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66, 1994 [PMID 7545954]
- Wooster R, Bignell G, Lancaster J, et al: Identification of breast cancer susceptibility gene BRCA2. Nature 378:789, 1995 [PMID 8524414]
- Scully R, Chen J, Plug A, et al: Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88:265, 1997 [PMID 9008167]
- Haber DA, Vogelstein B, Kinzler K: Wilms tumor. The Genetic Basis of Human Cancer. McGraw Hill Co, New York, 1998, pp 409-422
- Gnarra JR, Duan DR, Weng Y, et al: Molecular cloning of the von Hippel-Lindau tumor suppressor gene and its role in renal carcinoma. Biochim Biophys Acta 1242:201, 1996 [PMID 8603073]
- Polakis P: The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta 1332:F127, 1997
- Hahn SA, Schutte M, Hoque AT, et al: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271:350, 1996
- Hahn H, Wicking C, Zaphiropoulous PG, et al: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85:841, 1996 [PMID 8681379]
- McCormick F: Ras signaling and NF1. Curr Opin Genet Dev 5:51, 1995
- Gusella JF, Ramesh V, MacCollin M, et al: Neurofibromatosis 2: loss of merlin's protective spell. Curr Opin Genet Dev 6:87, 1996 [PMID 8791482]
- Parsons R: Phosphatases and tumorigenesis. Curr Opin Oncol 10:88, 1998
- Kolodner RD: Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends Biochem Sci 20:397, 1995
- Markowitz S, Wang J, Myeroff L, et al: Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268:1336, 1995 [PMID 7761852]
- Lynch HT, Smyrk T, Lynch JF: Molecular genetics and clinical pathology features of hereditary nonpolyposis colorectal carcinoma (Lynch syndrome): historical journey from pedigree anecdote to molecular genetic confirmation. Oncology 55:103, 1998
- Aaltonen LA, Salovaara R, Kristo P, et al: Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 338:1481, 1998 [PMID 9593786]
- Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 87:159, 1996 [PMID 8861899]
- Korsmeyer SJ: Regulators of cell death. Trends Genet 11:101, 1995
- Cleary ML, Smith SD, Sklar J: Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 47:19, 1986 [PMID 2875799]
- Rampino N, Yamamoto H, Ionov Y, et al: Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275:967, 1997 [PMID 9020077]
- Hayflick L, Moorhead PS: The serial cultivation of human diploid cell strains. Exp Cell Res 25:586, 1961
- Greider CW: Telomeres and senescence: the history, the experiment, the future. Curr Biol 8:R178, 1998
- Meyerson M, Counter CM, Eaton EN, et al: hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90:785, 1997 [PMID 9288757]
- Nakamura TM, Morin GB, Chapman KB, et al: Telomerase catalytic subunit homologs from fission yeast and human. Science 277:955, 1997 [PMID 9252327]
- Kolquist KA, Ellisen LW, Counter CM, et al: Expression of TERT in early premalignant lesions and a subset of cells in normal tissues. Nat Genet 19:182, 1998 [PMID 9620778]
- Liotta LA, Tryggvason K, Garbisa S, et al: Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 284:67, 1980 [PMID 6243750]
- Gunthert U, Hofmann M, Rudy W, et al: A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell 65:13, 1991 [PMID 1707342]
- Folkman J: Fighting cancer by attacking its blood supply. Sci Am 275(3):150, 1996
- O'Reilly MS, Holmgren L, Shing Y, et al: Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79:315, 1994
- O'Reilly MS, Boehm T, Shing Y, et al: Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88:277, 1997
- Boehm T, Folkman J, Browdar T, et al: Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 390:404, 1997 [PMID 9389480]
- Arap W, Pasqualini R, Ruoslahti E: Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279:377, 1998 [PMID 9430587]
- Kinsella AR, Smith D: Tumor resistance to antimetabolites. Gen Pharmacol 30:623, 1998 [PMID 9559310]
- Ling V: Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol 40:S3, 1997
- Grogan TM, Spier CM, Slamon SE, et al: P-glycoprotein expression in human plasma cell myeloma: correlation with prior chemotherapy. Blood 81:490, 1993 [PMID 8093668]
- Leith CP, Kopecky KJ, Godwin J, et al: Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group Study. Blood 89:3323, 1997
- Lowe S, Bodis S, McClatchey A, et al: Status and the efficacy of cancer therapy in vivo. Science 266:807, 1994 [PMID 7973635]
- Bischoff JR, Kirn DH, Williams A, et al: An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274:373, 1996 [PMID 8832876]
- Haber D, Fearon E: The promise of cancer genetics. Lancet 351(suppl II):1, 1998
- Sidransky D, Tokino T, Hamilton SR, et al: Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science 256:102, 1992 [PMID 1566048]
- Mao L, Schoenberg MP, Scicchitano M, et al: Molecular detection of primary bladder cancer by microsatellite analysis. Science 271:659, 1996 [PMID 8571131]
- Steiner G, Schoenberg MP, Linn JF, et al: Detection of bladder cancer recurrence by microsatellite analysis of urine. Nat Med 3:621, 1997 [PMID 9176487]
-
Ponder B: Genetic testing for cancer risk. Science
278:1050, 1997
-
Brennan JA, Mao L, Hruban RH, et al: Molecular assessment
of histopathological staging in squamous-cell carcinoma of the head and
neck. N Engl J Med 332:429, 1995 [PMID
7619114]
- Hyashi N, Arakawa H, Nagase H, et al: Genetic diagnosis identifies occult lymph node metastases undetectable by the histopathological method. Cancer Res 54:3853, 1994