Contenido del artículo
II ENFERMEDADES DEL SISTEMA NERVIOSO PERIFERICO
- Síntomas de los trastornos de los nervios periféricos
- Pruebas de laboratorio para los trastornos de los nervios periféricos
- Enfermedades de los nervios periféricos
- MONONEUROPATIAS CRANEALES
- MONONEUROPATIA DE LAS EXTREMIDADES
- Síndrome de tunel del carpo
- Neuropatía cubital
- Neuropatía del plexo braquial
- Neuropatía peronea
- Meralgia parestésica
- Plexopatía lumbosacra
- POLINEUROPATIAS
- Tratamiento de los síntomas de neuropatía
DR. COLIN H. CHALK, C.M.
DR. PETER JAMES DYCK
El sistema nervioso periférico (SNP) incluye todos los nervios craneales (excepto el primero y el segundo), las raíces nerviosas, los ganglios espinales, los nervios segmentarios, los plexos nerviosos y los nervios de las extremidades. El papel del SNP es simple pero vital: asociar el sistema nervioso central al ambiente externo y al medio interno del organismo.
Los nervios periféricos están compuestos por fascículos de endoneurio que contienen varios miles de axones y varias células de apoyo [ver figura 1]. Cada fascículo endoneural está envuelto por el perineurio (varias capas concéntricas de células semejantes a fibroblastos). Los troncos nerviosos están formados por varios fascículos endoneurales unidos entre sí por el epineurio (una malla laxa de colágena y fibroblastos). Existen dos redes compartamentales de vasos sanguíneos en los nervios, un sistema epineural de arteriolas y vénulas, que son tributarias de los vasos principales de las extremidades, y microvasos semejantes a capilares en el endoneurio. Las anastomosis entre las dos redes son extensas, lo que permite una irrigación abundante.
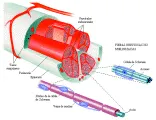
|
| Figura 1 |
| Estructura de un nervio periférico |
Cada axón es una extensión longitudinal de un cuerpo celular neuronal. La función y mantenimiento de los axones depende de la maquinaria bioquímica y genética del cuerpo celular. Un sistema bidireccional dependiente de energía de transporte axonal envía moléculas estructurales y de señales entre el cuerpo celular y el axón terminal. Los axones motores, sensoriales y autonómicos se mezclan en la mayoría de los nervios, pero los cuerpos celulares se agrupan en forma separada en la médula espinal, el tallo cerebral (motor), los ganglios de las raíces dorsales (sensorial) y los ganglios paravertebrales y somáticos (autonómicos).
Alrededor de un cuarto de los axones del SNP son mielinizados. La mielina es producida y mantenida por las células de Schwann, las células de apoyo más importantes en el endoneurio. Durante el desarrollo las células de Schwann elaboran grandes cantidades de membrana citoplásmica para formar una espiral alrededor de los axones. La membrana citoplásmica forma la capa lamelar de mielina. Una sola célula de Schwann mieliniza 0.2 a 1.8 mm de membrana citoplásmica a lo largo de un solo axón, lo que significa que la mielina que cubre toda la longitud de un solo axón es producto colectivo de varios cientos de células de Schwann consecutivas. Los axones no mielinizados, más numerosos, también se asocian íntimamente con las células de Schwann. El desarrollo normal de los axones y el desarrollo normal de las células de Schwann son interdependientes y se conocen muchos detalles de sus complejas interacciones.
Para los primeros anatomistas fue lógico dividir al sistema nervioso en central y periférico. Esa división no es arbitraria y varias diferencias entre el sistema nervioso periférico y el central tienen importancia clínica. Muy aparente es la simplicidad funcional del SNP. En esencia, es un sistema que trasmite las señales motoras, sensoriales y autonómicas entre el SNC y las estructuras somáticas. La correlación clínica consiste en el limitado repertorio de síntomas y signos producidos por malfunción del SNP. Es poco lo que se puede inferir sobre la causa del trastorno de un nervio periférico a partir solo de los síntomas y signos.
La segunda característica distintiva del SNP es su accesibilidad. A diferencia del cerebro y la médula espinal, que están bien protegidos por estructuras óseas, la mayoría de los nervios periféricos tienen la mayor parte de su curso a muy poca distancia por debajo de la piel. Esto simplifica el estudio directo de las características eléctricas e histológicas. La evaluación clínica de las enfermedades del SNP se ha facilitado mucho por los estudios de conducción nerviosa y la biopsia de nervio, que pueden realizarse prácticamente sin riesgo para el paciente.
La capacidad de regeneración difere entre el sistema nervioso periférico y el central. En el SNC maduro ocurre muy poco o ningun crecimiento de axones después de una lesión. Sin embargo, un nervio periférico lesionado con frecuencia puede regenerarse grandes distancias y restablecer conexiones funcionales. Por lo tanto, muchos trastornos de los nervios periféricos tienen posibilidad de recuperación de la función con el tratamiento.
Durante mucho tiempo se pensó que la diferencia en la capacidad regenerativa se relacionaba con alguna propiedad intrínseca de las neuronas del SNP. Sin embargo, evidencias recientes sugieren que el factor crítico se basa en los tejidos de apoyo. Si un haz de fibras nerviosas del SNC (v.gr., el nervio óptico o la médula espinal) es seccionado y después se reanastomosa de inmediato, existe poco crecimiento axonal. Sin embargo, si el extremo proximal de la misma lesión se anastomosa a un injerto de nervio periférico, el extremo axonal se extenderá varios milímetros a través del injerto de nervio periférico y puede incluso establecer conexiones funcionales con neuronas blanco del SNC.
Las bases celulares y moleculares de esta diferencia entre los tejidos de apoyo del SNP y del SNC no se conocen del todo. La célula de Schwann (exclusiva del SNP) parece ser la clave, aunque otras células (v.gr., fibroblastos y macrófagos) quizá participen también. Se piensa que la célula de Schwann y el medio extracelular del nervio periférico proporcionan un ambiente favorable para el crecimiento axonal, atrayendo y quizá guiando los muñones axonales. Dos principales clases de moléculas influyen en el crecimiento y desarrollo de los extremos axonales in vivo: los factores de crecimiento y las moléculas de adhesión. Algunas de éstas (v.gr., factor de crecimiento neural, N-cadherina y laminina) han sido bien caracterizadas. La comprensión de los eventos moleculares en la regeneración del SNP es un área de investigación activa dentro de la neurociencia que tiene importantes implicaciones biológicas y clínicas.1
Síntomas de los trastornos de los nervios periféricos
Los trastornos del SNP producen combinaciones de síntomas motores, sensoriales y autonómicos. Estos síntomas están determinados principalmente por la clase de fibras nerviosas afectadas (v.gr., motoras o sensoriales) y por la localización de las lesiones, más que por la etiología del proceso. Los síntomas motores incluyen debilidad (una molestia frecuente), atrofia muscular, fasciculaciones y calambres; estas dos últimas molestias pueden descubrirse solo después del interrogatorio específico. Los síntomas sensoriales incluyen la pérdida de ciertos tipos de sensación y la presencia de piquetes o sensación de quemadura. La propia descripción de la calidad y distribución de los síntomas sensoriales suele ser más reveladora que el examen mismo. Los síntomas sensoriales suelen ser desagradables o dolorosos. El dolor es un síntoma común en los padecimientos de los nervios periféricos y ocurre con frecuencia asociado con otros síntomas neuropáticos. Si el dolor es el único síntoma, su causa puede relacionarse con estructuras diferentes a los nervios periféricos. Los síntomas autonómicos son heterogéneos y con frecuencia se descubren por interrogatorio directo porque pueden no parecer relacionados para el paciente. Son comunes el mareo ortostático, la sudoración excesiva o disminuida, la impotencia o falla eyaculatoria y la alteración en la motilidad gastrointestinal (en especial el retraso en el vaciamiento gástrico).
Pruebas de laboratorio para los trastornos de los nervios periféricos
Con frecuencia se requieren estudios de laboratorio y gabinete para aclarar el origen de un trastorno del nervio periférico. Además de las pruebas usadas en la evaluación de otros tipos de enfermedad neurológica (v.gr., punción lumbar), se cuenta con técnicas para estudiar en forma directa la fisiología y la histología de los nervios periféricos.
ESTUDIOS DE CONDUCCION NERVIOSA
Los estudios de conducción nerviosa proporcionan indicación sobre el número de axones motores y sensoriales mielinizados en diversos nervios y de la velocidad de trasmisión del impulso en estas fibras. El procedimiento consiste en despolarizar un segmento corto de un nervio con una corriente eléctrica breve y registrar los potenciales de acción al propagarse por el nervio. Por este procedimiento se activan fibras tanto motoras como sensoriales y las respuestas motoras y sensoriales se distinguen entre sí al registrar una rama cutánea del nervio (sensorial) o un músculo inervado (motor).
Los estudios de conducción apoyan el diagnóstico de neuropatía y son una manera objetiva de vigilar la evolución de la enfermedad. Permiten también inferir si una enfermedad afecta principalmente los axones o sus vainas de mielina, una distinción importante en el diagnóstico diferencial de la polineuropatía. Si los estudios de conducción nerviosa muestran menor ampitud motora y sensorial pero velocidad de conducción conservada, el proceso patológico subyacente será probablemente pérdida o destrucción de axones. Por el contrario, si las amplitudes están relativamente conservadas y existe retraso marcado de la velocidad de conducción o bloqueo de la misma, la alteración podrá estar en la vaina de mielina.
ELECTROMIOGRAFIA CON AGUJA
La electromiografía con aguja es una investigación complementaria que suele realizarse al mismo tiempo que los estudios de conducción. El procedimiento consiste en registrar la actividad eléctrica en el músculo usando un electrodo de aguja insertado en el músculo. Los músculos se examinan en reposo y durante una contracción voluntaria lenta. Si la actividad eléctrica produce un patrón anormal, el electromiógrafo puede determinar si la debilidad del paciente es resulado de un músculo enfermo o de alteración en el nervio motor. En un paciente con neuropatía periférica la electromiografía es útil para detectar pérdida discreta de axones que puede no detectarse en los estudios de conducción nerviosa. La electromiografía también es útil para definir la localización precisa de la lesión en una mononeuropatía.
BIOPSIA DE NERVIO
La biopsia de nervio puede ser útil para el diagnóstico en pacientes seleccionados. La biopsia de nervio sural suele ser la preferida en la mayoría de los casos, ya que causa pérdida sensorial en un área pequeña del lado lateral del pie y esta suele ser bien tolerada. La biopsia de nervio puede proporcionar un diagnóstico específico (v.gr., vasculitis necrosante) pero con gran frecuencia da invormación menos específica que puede ser útil al combinarla con los otros datos clínicos. Además de la alteración en el número o tamaño de los axones, la alteración más encontrada en la biopsia de nervio es la inflamación, que suele relacionarse con alteración en la función inmunológica. En ocasiones puede diagnosticarse una enfermedad que afecte tanto el SNC como el SNP (v.gr., leucodistrofia metacromática).
El examen patológico adecuado de una biopsia de nervio requiere de técnicas disponibles solo en laboratorios especializados. Cuando no se dispone de estos servicios en la localidad, el paciente debe ser referido a una institución adecuada.
Enfermedades de los nervios periféricos
Es útil dividir las enfermedades del SNP en las que afectan nervios únicos (mononeuropatías) y las que afectan al SNP en forma difusa (polineuropatías). Una mononeuropatía afecta un nervio craneal o de una extremidad (ver adelante). En general, las mononeuropatías ocurren en personas por lo demás sanas, causan síntomas molestos pero no incapacidad, y suelen mejorar en forma espontánea con el tiempo.
MONONEUROPATIAS CRANEALES
Las neuropatías pueden aparecer en cualquier nervio craneal [ver tabla 1]. Los pares craneales séptimo (facial), quinto (trigémino) y tercero (oculomotor) son los asociados con la mayoría de las mononeuropatías craneales que se encuentran en la práctica clínica.
|
|||||||||||||||||||||||||||||||||||||||
|
Neuropatia facial
La neuropatía facial idiopática aguda (parálisis de Bell) es la causa más común de neuropatía craneal, con una incidencia anual de alrededor de 25 por 100,000 personas. El principal síntoma es la debilidad facial unilateral que comienza en forma súbita y suele ser precedida o acompañada por dolor detrás del oído. En algunos pacientes pudo haber ocurrido infección leve de las vías respiratorias superiores en las 2 semanas previas. La debilidad facial suele alcanzar su máximo a las 24 horas de inicio y es de tipo motoneurona inferior, afectanto la porción tanto superior como inferior de la cara. Algunos pacientes sufren alteración del sentido del gusto en la mitad de la lengua e hiperacusia. Suelen referir además sensación de adormecimiento en la cara, pero no se encuentra pérdida sensorial.
Cuando la historia y el examen físico son típicos de parálisis de Bell y el proceso es leve y no progresivo no se requieren estudios. Los casos severos o que progresan deben referirse a un neurólogo para confirmar el diagnóstico y decidir si se requiere intervención. En ocasiones la sarcoidosis, la enfermedad de Lyme y el herpes zoster se manifiestan por neuropatía craneal y puede ser adecuado realizar pruebas para excluir estos padecimientos. Cuando el examen neurológico revela más que la neuropatía facial unilateral o cuando la debilidad ha evolucionado en semanas o meses debe pensarse en un diagnóstico alternativo, como un tumor del ángulo pontocerebeloso o una lesión del tallo cerebral.
La historia natural de la parálisis de Bell no tratada suele ser favorable: el 85 porciento de los pacientes se recuperan en 1 año. Sin embargo, algunas manifestaciones clínicas se asocian con mal pronóstico, incluyendo edad avanzada, parálisis facial completa y la presencia de hiperacusia o alteración en el gusto. El ocasiones se desarrolla espasmo hemifacial, un síndrome de contracciones involuntarias, breves y no dolorosas de los músculos faciales después de la parálisis de Bell y de otros tipos de neuropatía facial.
Aunque existe debate sobre si la parálisis de Bell es un fenómeno autoinmune posinfeccioso o una infección viral directa del nervio facial, existe acuerdo sobre que la patogenia incluye inflamación del nervio facial. De acuerdo con ello, la parálisis de Bell suele tratarse con esteroides. Varios estudios han sugerido que el tratamiento esteroideo acelera la recuperación, pero no existen estudios satisfactorios controlados con placebo. Es adecuado administrar un curso breve y de disminución gradual de esteroides en los pacientes que se atienden en los primeros días de inicio (v.gr., prednisona 60 mg/día durante 5 días, disminuyendo hasta cero en los siguientes 10 días). La posibilidad de infección viral directa del nervio facial ha sido apoyada por un estudio japonés reciente en el que se encontró ADN del virus herpes simple tipo 1 en muestras de nervio facial de 11 de 14 pacientes con parálisis de Bell, pero no en pacientes con otras causas de neuropatía facial, incluyendo herpes zoster.2 Un estudio controlado de aciclovir más prednisona contra prednisona sola encontró mejoría modesta pero significativa en la evolución clínica del grupo tratado con aciclovir.3 Con base en la evidencia actual, la combinación de aciclovir y prednisona puede considerarse razonable para la mayoría de los pacientes con parálisis de Bell, a pesar de que la historia natural de esta condición suele ser benigna, independientemente del tratamiento. Es importante evitar la desecación corneal con un parche ocular en los pacientes que no pueden cerrar el ojo por completo. En alguna ocasión se usó la descompresión del nervio facial, pero el procedimiento ha sido prácticamente abandonado por que no se demostró su eficacia. Las inyecciones de toxina botulínica son útiles para tratar el espasmo hemifacial.
Neuralgia del trigémino (tic doloroso)
La neuralgia del trigémino, o tic doloroso, es un síndrome facial doloroso de personas de edad media y ancianos en los que ocurre dolor intenso y paroxístico varias a docenas de veces al día. El dolor se describe como una descarga eléctrica intensa, dura segundos y se localiza en la distribución del trigémino, por lo general en los territorios maxilar y mandibular. Los paroxismos de dolor pueden ser desencadenados por estímulos tactiles leves del lado afectado de la cara, como rasurarse, lavarse o masticar. Entre los episodios de dolor los pacientes pueden tener dolor sordo, pero no adormecimiento ni parestesia. La neuralgia del trigémino puede confundirse con el dolor dental. Un síndrome semejante pero mucho más raro, la neuralgia del glosofaríngeo, produce paroxismos de dolor faríngeo.
El examen de los nervios craneales suele ser difícil en los pacientes con neuralgia del trigémino porque despierta el dolor. Sin embargo, no se encuentra pérdida sensorial u otros signos. Los nervios craneales y otros signos neurológicos obligan a investigar lesiones estructurales del nervio o del núcleo del trigémino, así como lesión del tallo cerebral. La esclerosis múltiple es el diagnóstico más probable en esta situación, pero suele estar bien establecido antes de que ocurran episodios de neuralgia del trigémino.
La causa de la neuralgia del trigémino es dudosa, aunque muchos investigadores la atribuyen a la compresión o distorsión del nervio trigémino por pulsaciones de una arteria cerebelosa superior ectásica. Ocurren remisiones espontáneas, pero que rara vez son duraderas.
La base del tratamiento es la carbamacepina, 200 a 1,200 mg/día. Una alternativa es el baclofén, 30 a 60 mg por día. Si la neuralgia del trigémino se vuelve refractaria a los medicamentos puede intentarse un enfoque quirúrgico, que suele ser útil. La rama apropiada del trigémino puede inyectarse con alcohol o fenol, o puede hacerse una lesión percutánea en el ganglio del trigémino. Sin embargo, con estos procedimientos el paciente cambia el dolor por diversos grados de anestesia, lo que no siempre es preferible. La descompresión microvascular para aliviar el posible efecto de la arteria cerebelosa superior suele ser muy eficaz en manos de cirujanos expertos, pero el procedimiento requiere de una craneotomía de fosa posterior.
Neuropatía oculomotora
Las parálisis aisladas del tercer par no son comunes, pero implican un diagnóstico diferencial difícil entre causas graves y benignas. El inicio es súbito y la diplopia y la cefalea suelen ser los síntomas habituales. La diplopia es tanto horizontal como vertical y la relación entre las dos imágenes cambia según la dirección de la mirada. La cefalea puede ser retro-orbitaria o difusa. La mayoría de los pacientes tienen ptosis y algunos dilatación pupilar.
La principal consideración en un paciente con parálisis aislada del tercer par son la compresión por una masa que se expande (en especial un aneurisma de la arteria comunicante posterior) y una lesión llamada microvascular, en la que se supone que la parálisis oculomotora es isquémica y causada por un trastorno en la irrigación al nervio. Una vez que es claro que los únicos signos son los de una parálisis del tercer par, el examen puede ayudar a distinguir entre una lesión compresiva y una lesión microvascular. Si existe parálisis pupilar es más probable que la lesión sea compresiva y está indicado realizar una tomografía computada y una angiografía cerebral. La parálisis del tercer par que respeta la pupila es más probable que sea microvascular, los pacientes típicamente son ancianos, diabéticos e hipertensos. Sin embargo, en un paciente joven que no es diabético ni hipertenso está justificado realizar una investigación neurorradiológica aunque la pupila esté respetada. La angiografía por resonancia magnética y la tomografía computada, que son no invasivos, han sustituido cada vez más a la angiografía cerebral convencional.
MONONEUROPATIA DE LAS EXTREMIDADES
Las mononeuropatías de las extremidades son comunes. La compresión mecánica del nervio en una localización vulnerable suele suponerse como causa, pero esto no siempre se demuestra en forma fehaciente. Otros padecimientos raros, como un tumor nervioso, pueden causar un cuadro clínico idéntico. Debe considerarse el síndrome autosómico dominante de predisposición hereditaria a la parálisis por presión en el paciente con mononeuropatías compresivas recurrentes o cuando el traumatismo precipitante es trivial comparado con el daño resultante.
Síndrome de tunel del carpo
El síndrome de tunel del carpo (STC) es causado por la compresión de la porción distal del nervio mediano al atravesar el tunel localizado en la muñeca y formado por los huesos del carpo en la porción inferior y el ligamento del carpo arriba. El paciente refiere adormecimiento de la mano (v.gr., sensación de piquetes, pérdida sensorial o ambas) que es molesta o dolorosa. Las parestesias suelen localizarse en la distribución sensorial del mediano, pero algunos pacientes refieren afección de todos los dedos o molestia del antebrazo. Los síntomas característicamente son episódicos y ocurren por la noche (despertando al paciente) y durante las actividades en las que las muñecas se mantienen en flexión o extensión, como al tejer o sostener un volante. La sacudida fuerte de las manos suele producir alivio inmediato y es muy sugestiva de STC. Las parestesias pueden dificultar el uso de la mano en forma adecuada, pero es rara la verdadera debilidad. Cuando existe menor fuerza de prehensión relacionada a debilidad de los músculos tenar, puede interferir con actividades como sostener una pluma. En algunos pacientes (en especial los ancianos) las molestias sensoriales pueden ser mínimas y predominar los síntomas de debilidad tenar.
En la mayoría de los pacientes el STC parece desarrollarse por angostamiento congénito del tunel. Ciertas ocupaciones que requieren de flexión y extensión repetitiva del tunel (v.gr., carpintería, costura y uso de un teclado de computadora) pueden predisponer al STC. El síndrome es común durante el embarazo (quizá por la retención generalizada de líquido) y puede ocurrir después de lesiones como una fractura de Colles. En ocasiones ocurre STC en el contexto de una enfermedad sistémica que disminuye el espacio libre en el tunel, como la artritis reumatoide, el mixedema, la amiloidosis y la acromegalia. Los pacientes con polineuropatía diabética y de otro tipo pueden tener STC y otras mononeuropatías, pero esta teoría no se ha demostrado del todo. En los pacientes con diabetes la mayor rigidez y volumen del ligamento del carpo puede ser un factor contribuyente importante.
El examen puede revelar deficiencias sensoriales o motoras en la distribución del mediano, pero con frecuencia es normal, en especial en pacientes jóvenes con síntomas intermitentes. La percusión sobre el nervio mediano en la muñeca puede producir parestesia temporal en los dedos inervados por el mediano (signo de Tinel). Sostener la muñeca en flexión o extensión forzada por 1 a 2 minutos puede precipitar los síntomas sensoriales del paciente, que se alivian con prontitud colgando el brazo en forma lateral sin angular la muñeca (signo de Phalen). Ninguno de estos signos son totalmente específicos o sensibles.
Los estudios de conducción nerviosa suelen apoyar el diagnóstico clínico de STC. El principio esencial consiste en demostrar retraso en la conducción del nervio mediano en el tunel del carpo comparado con los nervios cubital o radial vecinos. Los estudios de conducción nerviosa tienen alta sensibilidad diagnóstica para el STC, pero pueden ser normales en los casos leves y permanecen anormales incluso después de la cirugía exitosa.
Los pacientes con síntomas leves y sin deficiencias pueden solo necesitar que se les tranquilice. Suele ser eficaz una férula que sostenga la muñeca en posición neutral para los síntomas nocturnos. Algunos pacientes mejoran con inyecciones periódicas de esteroides en el ligamento del carpo. La resección quirúrgica del ligamento carpiano (un procedimiento de bajo riesgo) se usa mucho para el STC y suele causar mejoría importante y duradera en más del 90 porciento de los pacientes. La cirugía es adecuada para los pacientes con síntomas incapacitantes y que no responden a los tratamientos quirúrgicos, así como para los que tienen deficiencia motora o sensorial progresiva.
Neuropatía cubital
La neuropatía cubital suele ser causada por lesión mecánica en el codo, en donde el nervio yace subcutáneo y descansa en el piso del surco epicondíleo. Distal al codo el nervio puede comprimirse al pasar bajo la aponeurosis del flexor cubital del carpo hacia el compartimiento muscular profundo del antebrazo. En la muñeca el nervio nuevamente se vuelve relativamente superficial y puede comprimirse por el esfuerzo excesivo con las manos, como cuando se realiza ciclismo de distancias largas.
Los pacientes con neuropatía cubital presentan síntomas y signos de varios grados. Los pacientes observadores notarán que la alteración sensorial afecta la porción medial del dedo anular, además del dedo pequeño y la parte medial de la mano. Cuando existe debilidad se confina a la mano. La mano puede parecer débil en general o pueden existir molestias específicas, como dificultad para sostener una pluma o extender y separar los dedos.
La debilidad y atrofia se identifican principalmente en el primer múculo interóseo dorsal (entre el pulgar y el índice) y en los músculos hipotenar. Es rara la pérdida completa de la sensación cubital, pero puede estar alterada. La comparación entre los lados medial (cubital) y lateral (mediano) del dedo anular proporciona evidencia convincente de una neuropatía cubital. La producción de piquetes o sensación eléctrica en la mano con la manipulación suave del nervio en el codo apoya que éste sea el sitio de la patología. Las pruebas de electrodiagnóstico son útiles en la neuropatía cubital tanto para confirmar el diagnóstico como para localizar el sitio preciso de lesión.
En casos raros la neuropatía cubital puede ser causada por compresión en la porción superior del brazo o en la mano. Debido a la disposición de las ramas motoras y sensoriales en la mano y la muñeca, la compresión cubital en la mano solo afecta ramas motoras. Esto causa debilidad de la mano pero no síntomas de dolor o sensoriales y puede confundirse con la fase inicial de una enfermedad de motoneurona. Este tipo de neuropatía cubital es causada por esfuerzo en las manos, como el producido al usar una andadera de cuatro puntos de apoyo o con el ciclismo prolongado.
El tratamiento apropiado de la neuropatía cubital en el codo no es claro. Aunque el padecimiento es frecuente, existe muy poca información sobre su historia natural. La mejoría espontánea es frecuente, en especial en pacientes con un episodio precipitante claro de compresión. En los pacientes en los que el traumatismo leve y repetitivo del nervio es la causa la neuropatía cubital mejora evitando el apoyo sobre los codos.
Se usan varios procedimientos quirúrgicos en el tratamiento de la neuropatía cubital a nivel del codo. En pacientes seleccionados que tienen síntomas y signos progresivos a pesar del tratamiento conservador debe pensarse en la cirugía, pero sus méritos en comparación con los de los procedimientos no quirúrgicos no se han estudiado con cuidado.
Neuropatía del plexo braquial
La neuropatía del plexo braquial (también conocida como amiotrofia neurálgica y síndrome de Parsonage-Turner) es poco frecuente pero quizá también subdiagnosticada. La historia es característica. El paciente desarrolla dolor severo semejante al de la bursitis en el hombro sin un factor precipitante aparente. Después de varios días a 2 semanas el dolor cede y el paciente nota la debilidad en el brazo. Con más frecuencia se afectan los músculos de la cintura escapular, pero también pueden afectarse la mano o el antebrazo. En ocasiones la debilidad es focal o en parches y puede haber pérdida sensorial en una distribución en charretera, pero esta no es una característica muy notoria.
Los pacientes se recuperan en forma gradual y el 90 porciento tiene muy poca limitación o ninguna 1 a 2 años después. En algunos pacientes se afectan los dos brazos en forma simultánea o secuencial y algunos enfermos tienen episodios recurrentes. La neuropatía del plexo braquial es más común en varones jóvenes y de edad media, pero puede afectar ambos sexos y todos los grupos de edad. Las biopsia del plexo braquial han demostrado inflamación linfocítica florida y el trastorno parece ser de origen inmune.4 Sin embargo, el tratamiento inmunomodulador no ha demostrado ser benéfico. Existe también una forma autosómico dominante de neuropatía del plexo braquial que se caracteriza por episodios recurrentes de dolor y debilidad en el hombro.
Neuropatía peronea
La neuropatía peronea suele ser resultado de lesión del nervio peroneo en el punto en el que rodea la cabeza del peroné, pasando de la fosa poplítea al compartimiento anterior de la pierna. Las ramas peroneas distales son superficiales a nivel del tobillo, en donde en ocasiones son comprimidas por zapatos muy estrechos. Los pacientes con neuropatía peronea en la rodilla suelen presentar pie caído unilateral por debilidad de los dorsiflexores del tobillo.Sin embargo, el pie caído también puede ser causado por una radiculopatía L5, una plexopatía lumbosacra, una neuropatía ciática proximal o una lesión en la corteza motora cerebral contralateral. La radiculopatía L5, el diagnóstico diferencial más común, es sugerida por la presencia de lumbalgia que irradia a la pierna y por debilidad de los invertores del pie, los abductores de la cadera, los dorsiflexores del tobillo y los extensores del primer ortejo. Las pruebas electrodiagnósticas pueden ser especialmente útiles para localizar la lesión en el paciente con pie caído.
Suele existir historia de traumatismo obvio (v.gr., fractura del peroné) en los pacientes con neuropatía peronea de la rodilla. La compresión durante los estados de alteración de la conciencia (v.gr., durante la anestesia y el coma) es otra causa común. La herniación de un quiste de Baker puede comprometer el nervio. En los pacientes sin estos factores precipitantes la neuropatía peronea a nivel de la rodilla suele atribuirse al sentado habitual con la pierna cruzada, una explicación plausible aunque difícil de probar.
El tratamiento de la neuropatía peronea a nivel de la rodilla suele ser conservador. En la mayoría de los pacientes que han sufrido un traumatismo al nervio ocurre mejoría con el tiempo, en especial cuando existe cierta función residual del nervio peroneo distal. En los traumatismos severos el nervio puede seccionarse por completo, caso en el que la evolución es mala y la anastomosis quirúrgica constituye la mejor opción para la recuperación. Cuando no existe una causa aparente para la neuropatía peronea los pacientes deben evitar sentarse con la pierna cruzada y puede ocurrir cierta mejoría. Independientemente de la causa, debe intentarse una férula de tobillo pie para compensar el pie caído; la mayoría de los pacientes encuentran que este aditamento mejora mucho su capacidad para caminar [ver adelante, Tratamiento de la neuropatía sintomática].
Meralgia parestésica
La meralgia parestésica se caracteriza por síntomas sensoriales en la distribución del nervio cutáneo lateral del muslo. Los pacientes reportan cierta combinación de pérdida de la sensibilidad, piquetes e hipersensibilidad sobre la región anterolateral del muslo. Los límites del área anormal suelen estar bien definidos, sin alteraciones motoras o de reflejos asociadas. La meralgia parestésica suele atribuirse a la compresión del nervio cutáneo lateral por el ligamento inguinal en su paso desde el retroperitoneo hacia la región anterior del muslo. El síndrome es más común en las personas obesas, quizá por mayor estrés mecánico sobre el ligamento inguinal. En casos raros el nervio cutáneo lateral se lesiona durante una cirugía o puede ser presionado por ganglios inguinales crecidos u otras masas. La mayoría de los pacientes solo requieren ser tranquilizados porque los síntomas ceden con el tiempo. En algunos pacientes con síntomas persistentes y molestos puede intentarse el tratamiento con medicamentos como la amitriptilina. La exploración quirúrgica de la región inguinal es un último recurso.
Plexopatía lumbosacra
La plexopatía lumbosacara se parece a la neuropatía del plexo braquial. La historia típica de afección del plexo lumbosacro comienza con dolor en la región anterior del muslo, por lo general severo y continuo, que dura varias semanas. Al ceder el dolor aparece debilidad, principalmente en el cuadríceps y flexores de la cadera. Con frecuencia la debilidad no causa síntomas hasta que el paciente se cae, dando la impresión equivocada de un evento agudo. Puede ocurrir atrofia impresionante del cuadríceps y el reflejo rotuliano está ausente. Los signos sensoriales son mínimos. En ocasiones los signos y síntomas iniciales afectan solo la parte lumbar o sacra del plexo, pero existe la tendencia a que la plexopatía lumbosacra afecte todo el plexo y el lado contralateral. la recuperación muy gradual es la regla.
La plexopatía lumbosacra afecta característicamente varones de edad media y ancianos. Los pacientes con frecuencia tienen diabetes (caso en el que se usan los términos de amiotrofia diabética, neuropatía diabética femoral y neuropatía diabética proximal) aunque puede ocurrir un síndrome idéntico en los pacientes no diabéticos. Las pruebas de electrodiagnóstico son útiles para apoyar el diagnóstico. Los estudios de imagen permiten excluir invasión maligna del plexo lumbar, hematomas retroperitoneales y abscesos del psoas.
La patogenia de la plexopatía lumbosacra es motivo de discusión. Durante mucho tiempo se apoyó una base isquémica, principalmente porque ocurre con frecuencia en pacientes con complicaciones microvasculares de la diabetes. Sin embargo, existen cada vez más evidencias por los estudios de biopsia de un proceso inflamatorio, quizá autoinmune.5 No existen evidencias claras de que la inmunomodulación sea eficaz en la plexopatía lumbosacara, pero en la actualidad se realizan estudios controlados al respecto. El dolor que dura por más de algunas semanas puede mejorar con medicamentos como la amitriptilina.
POLINEUROPATIAS
Por lo general no es difícil determinar que un paciente tiene una polineuropatía. Sin embargo, debido a que existen alrededor de cien causas de polineuropatía que producen síntomas y signos semejantes, puede ser muy difícil llegar a un diagnóstico específico. El diagnóstico diferencial puede simplificarse con las siguientes preguntas:
1.¿Los síntomas y signos tienen un patrón diferente al de una polineuropatía sensorimotora simétrica? Varios síndromes distintivos, a diferencia de las polineuropatías sensorimotoras simétricas, tienen escasos diagnósticos diferenciales. Son ejemplos la mononeuropatía múltiple (también conocida como mononeuritis múltiple), la neuropatía sensorial atáxica, la neuropatía autonómica pura o predominante y la polineuropatía de inicio súbito [ver tabla 2].
|
|
* Por lo general se manifiesta como polineuropatía simétrica |
2.¿Los estudios de conducción nerviosa sugieren desmielinización? Después de la historia clínica y el examen físico, los estudios de conducción nerviosa son el siguiente paso lógico. Si la velocidad de conducción es muy lenta o existen otras evidencias electrofisiológicas que sugieren desmielinización el diagnóstico diferencial es fácil [ver tabla 3].
|
|
|
3.¿La polineuropatía es adquirida o hereditaria? Esta es una consideración importante en todos los pacientes con polineuropatía crónica simétrica. La neuropatía hereditaria es la causa que más frecuentemente se pasa por alto de polineuropatía no diagnosticada.6 El pie cavo y los dedos en martillo pueden ser claves importantes para el diagnóstico de neuropatía hereditaria y deben siempre buscarse en forma específica. Las neuropatías hereditarias suelen ser leves, no incapacitantes y de muy lenta progresión. Por lo tanto, con frecuencia no están presentes sino hasta la mitad de la vida o más adelante y con frecuencia pasan desapercibidas en la familia [ver tabla 4].
|
||||||||||||||||||||
|
La investigación subsecuente de las neuropatías axonales, sensorimotoras y simétricas puede guiarse por información colateral en la historia. Si existe posibilidad de una neuropatía hereditaria deben examinarse los familiares en primer grado. La causa de una neuropatía adquirida puede ser un medicamento [ver tabla 5] o una enfermedad concomitante, como diabetes mellitus e insuficiencia renal crónica como las más comunes. En ocasiones la neuropatía puede ser el dato de presentación de una vasculitis sistémica o una neoplasia oculta.
|
|
|
En los pacientes adultos las pruebas de laboratorio justificadas incluyen una glucemia en ayuno, concentración de hemoglobina glucosilada, nivel de creatinina en suero, biometría hemática, radiografía de tórax (en los fumadores), velocidad de sedimentación, factor reumatoide, anticuerpos antinucleares y en pacientes mayores de 50 años inmunoelectroforesis de las proteínas de plasma y orina. La biopsia de nervio puede ser valiosa, pero solo si se busca información específica. No es una prueba de rutina y rara vez es útil, incluso cuando se realiza como último recurso.
Polineuropatía diabética
La polineuropatía diabética es común en pacientes con diabetes. De los varios tipos de neuropatía, con mucho la más común es una neuropatía sensorimotora simétrica distal conocida como polineuropatía diabética. Es difícil calcular la incidencia y severidad de la polineuropatía diabética por (1) la forma de definir la enfermedad, (2) las poblaciones que se han estudiado y (3) los esfuerzos para asegurar que se excluyan otras causas de neuropatía. En un estudio poblacional prospectivo se encontró polineuropatía diabética en el 54 porciento de los pacientes con diabetes mellitus insulinodependiente y en el 45 porciento de los pacientes con diabetes mellitus no insulinodependiente.7 Sin embargo, ocurrió polineuropatía sintomática en solo el 15 porciento de la cohorte y ninguno de los pacientes tenía deficiencia neurológica limitante. En este y otros estudios grandes, la prevalencia de la polineuropatía diabética aumentó con la duración de la enfermedad y existió intensa correlación entre la presencia de polineuropatía, retinopatía y nefropatía. Un corolario práctico e importante de estas observaciones es que el diagnóstico de polineuropatía diabética en un paciente con diabetes recién diagnosticada pero sin otras complicaciones es probable que sea incorrecto.
La polineuropatía diabética tiene la distribución clásica llamada de guante y calcetín, por lo general combinando pérdida de sensibilidad y sensación desagradable de adormecimiento o quemadura. La pérdida de sensibilidad en los pies y dedos y la debilidad leve en los pies y tobillos son típicas. Es de esperar que la polineuropatía diabética empeore un poco con los años,8 aunque esta progresión puede depender de cómo se trate la diabetes. No son frecuentes las deficiencias neurológicas muy limitantes ni el empeoramiento rápido, por lo que si existen obligan a investigar otras causas.
La patogenia de la polineuropatía diabética sigue siendo motivo de controversia. Una teoría popular es la mayor actividad de la vía poliol secundaria a la hiperglucemia crónica que causa acúmulo de sorbitol y depleción de mio-inositol en el nervio. A su vez, la reducción de mio-inositol parece producir menor actividad de la Na+-K+-ATPasa, causando alteración en la conducción nerviosa y alteraciones estructurales en el axón. En animales diabéticos algunas de estas alteraciones pueden demostrarse y corregirse inhibiendo la vía del poliol, pero en los nervios de humanos con diabetes el contenido de mio-insitol es normal.9 En estudios clínicos en humanos se han observado pequeñas mejoras electrofisiológicas durante el tratamiento con inhibidores de la aldosa reductasa, que suprimen la actividad de la vía poliol.10 Aún es motivo de controversia si este tratamiento es útil.
Una hipótesis alternativa para la polineuropatía diabética es la de isquemia-hipoxia.11 En el nervio y otros tejidos de pacientes diabéticos ocurre degeneración de pericitos y reduplicación de la lámina basal capilar, que puede implicar a los microvasos en la patogenia de complicaciones como la neuropatía. En los diabéticos puede demostrarse reducción en el flujo sanguíneo nervioso y en la tensión endoneural de oxígeno, y se logra corrección parcial de las alteraciones de conducción nerviosa por medio de oxígeno suplementario.12 En biopsias de pacientes con polineuropatía diabética se ha demostrado que el grado de pérdida de fibras nerviosas es muy variable entre los fascículos, patrón que se observa en las neuropatías con una base isquémica bien establecida, como la neuropatía vasculítica. En la actualidad la hipótesis hipóxica parece ser la más aceptada, pero no necesariamente excluye la participación de la vía poliol u otras alteraciones metabólicas.
Los pacientes con polineuropatía diabética deben ser aconsejados sobre un buen cuidado en los pies, inspección diaria cuidadosa de las superficies plantares para detectar pronto las lesiones leves y evitar complicaciones mayores como úlceras plantares u osteomielitis. En ocasiones se requiere manejo específico del dolor neuropático.
El efecto benéfico del buen control de la glucemia en la polineuropatía diabética es cada vez más evidente. Después de 5 años de seguimiento en el Estudio sobre Control y Complicaciones de la Diabetes, la prevalencia de polineuropatía fue de 5 porciento en los pacientes con un control riguroso de la glucemia comparado con 13 porciento en los que recibieron tratamiento convencional.13 El seguimiento de una cohorte de receptores de trasplantes de páncreas reportó mejoría significativa en los parámetros de conducción nerviosa y quizá mejoría modesta en el examen clínico; en los pacientes controles que no recibieron trasplante estos parámetros empeoraron durante el mismo periodo.14 Por lo tanto, el control óptimo de la glucemia parece disminuir el riesgo de desarrollar polineuropatía diabética. En la polineuropatía establecida puede tener un efecto benéfico, pero aún no se demuestra si ocurre mejoría clínica significativa durante varias décadas. La esperanza de contar con fármacos adicionales para prevenir o aliviar la neropatía diabética ha originado numerosos estudios clínicos que han evaluado medicamentos como los inhibidores de la aldosa reductasa, la acilcarnitina, la aminoguanidina (que disminuye los productos terminales de la glucosidación avanzada), el aceite de flor de primavera (una fuente de ácido linoleico), el ácido alfa-lipoico (un depurador de radicales libres de oxígeno) y factores de crecimiento neural. A la fecha algunos de estos estudios han sugerido beneficios modestos, pero problemas en el diseño de los estudios (v.gr., duración del tratamiento y si los resultados fueron significativos desde el punto de vista clínico) han evitado que se forme un consenso sobre el uso práctico de estos agentes.15
Otras neuropatías diabéticas
Las otras variedades de neuropatía diabética suelen ocurrir sobre el fondo de una polineuropatía diabética. Dos mononeuropatías (la oftalmoplegia diabética y la neuropatía diabética proximal) pueden ocurrir también en pacientes diabéticos [ver antes, Neuropatía oculomotora y Plexopatía lumbosacra].
Neuropatía diabética autonómica Se encuentra cierto grado de neuropatía autonómica diabética en la mayoría de los pacientes con polineuropatía diabética, aunque en algunos pacientes predominan los síntomas y signos autonómicos. La hipotensión ortostática, la menor motilidad gastrointestinal (incluyendo la gastroparesia) y el bloqueo de síntomas de advertencia simpática ante la hipoglucemia son problemas de manejo importantes. Algunos síntomas autonómicos pueden tratarse en forma eficaz, por ejemplo, la gastroparesia mejora con metoclopramida y el ortostatismo con medias elásticas y expansión de volumen.
Neuropatía diabética troncal La neuropatía diabética troncal es un síndrome sensorial doloroso que afecta uno o varios dermatomas torácicos, por lo general en forma unilateral. Desde el punto de vista anatómico, el proceso es una radiculopatía torácica con desarrollo súbito de hipersensibilidad tactil y pérdida sensitiva en la distribución de un dermatoma. Por lo general puede demostrarse afección motora por electromiografía y en ocasiones abultamiento asimétrico del abdomen por debilidad unilateral de los músculos. El principal diagnóstico diferencial es la radiculopatía por herpes zoster, que suele distinguirse por la erupción vesicular en la piel. La radiculopatía diabética troncal es autolimitada, aunque los síntomas pueden persistir por semanas. El manejo se basa en administración de analgésicos suplementados por amitriptilina.
Otros trastornos metabólicos
Se desarrolla polineuropatía en hasta el 60 porciento de los pacientes con insuficiencia renal crónica. El riesgo de desarrollar neuropatía urémica se relaciona con la duración y severidad de la insuficiencia renal. Se observa reducción en la velocidad de conducción cuando la depuración de creatinina baja a menos del 10 porciento del normal, aunque la velocidad de conducción no correlaciona mucho con los síntomas. La neuropatía urémica es sensorimotora y los síntomas y signos son principalmente distales. Una vez establecida tiende a empeorar con lentitud y los principales síntomas son disestesias desagradables en los pies. Es rara la debilidad motora incapacitante. El principal dato patológico en la neuropatía urémica consiste en degeneración axonal, que es más abundante en las partes distales del SNP. Es posible que alguna o varias sustancias neurotóxicas excretadas normalmente por los riñones alteren la función del cuerpo neuronal o del axón, pero no se conocen los detalles al respecto. Tanto la diálisis como el trasplante renal suelen ser benéficos. La diálisis puede prevenir, estabilizar y con frecuencia mejorar la neuropatía. Los efectos del trasplante renal pueden ser francos, y es de esperar que incluso la neuropatía urémica severa mejore en los meses siguientes al trasplante. La disponibilidad de estos tratamientos ha hecho que la neuropatía urémica sintomática sea relativamente poco frecuente.
En ocasiones ocurre polineuropatía axonal distal leve en pacientes con hipotiroidismo, acromegalia o policitemia. Estas neuropatías no son limitantes pero los síntomas sensitivos pueden ser molestos. Los pacientes en unidades de cuidados intensivos con síndrome de disfunción orgánica múltiple y sepsis en ocasiones desarrollan polineuropatía axonal severa. Se desconoce la patogenia de esta condición, conocida como polineuropatía de la enfermedad grave.16 Este tipo de neuropatía se sospecha cuando el paciente de la UCI no puede ser destetado del ventilador a pesar de una función cardiopulmonar adecuada o cuando existe debilidad de las extremidades en pacientes alerta. Debido a la dificultad para examinar el SNP en forma adecuada en el paciente grave y ya que la polineuropatía de la enfermedad grave es un diagnóstico de exclusión, puede ser difícil establecer el diagnóstico con claridad. La miopatía asociada a enfermedad grave puede dar un cuadro clínico semejante, en especial en pacientes tratados con dosis altas de esteroides y bloqueadores neuromusculares de acción prolongada (v.gr., vecuronio).17 Algunos datos sugieren que la neuropatía de la enfermedad grave mejora en pacientes que sobreviven al padecimiento de base.
Polineuropatías hereditarias
Las polineuropatías hereditarias son comunes y con frecuencia pasan desapercibidas, en especial en adultos.6 Los pacientes suelen estar relativamente asintomáticos por muchos años y, por lo tanto, no solicitar ayuda médica sino hasta la edad adulta o después. La historia de síntomas que progresan en forma muy lenta durante años es sugestiva de una causa hereditaria. En la polineuropatía hereditaria el paciente refiere adormecimiento (hiposensibilidad), mientras que la presencia de piquetes orienta más hacia una causa adquirida. Sin embargo, la sensación de ardor en los pies es inespecífica y ocurre en las polineuropatías tanto hereditarias como adquiridas. Como regla, las neuropatías hereditarias comienzan y progresan en forma simétrica, la progresión asimétrica sugiere una causa adquirida.
Cuando se sospecha polineuropatía hereditaria, probar el diagnóstico puede ser considerablemente difícil. Rara vez es útil preguntar al paciente sobre la historia familiar de neuropatía. El interrogatorio debe ser más general, como si alguien tiene molestias en los pies, deformidad en los mismos (especialmente arcos muy altos) o requiere zapatos especiales, bastón o tirantes. La historia familiar negativa no excluye el diagnóstico. Por su cronicidad y poca severidad, es frecuente que el trastorno pase desapercibido en otros miembros de la familia. En los csos recesivos, por definición ningún padre está afectado y los hijos pueden tampoco estarlo. El siguiente paso útil consiste en examinar a los familiares en primer grado, en especial los que tienen historia de problemas con los pies o el caminar. Aunque este procedimiento toma tiempo, tiene mejor relación costo-beneficio que realizar una batería de laboratorio que será negativa.
El conocimiento sobre los defectos genéticos específicos relacionados con las diversas neuropatías hereditarias está creciendo con rapidez,18 pero la clasificación clínica sigue siendo importante. En forma tradicional, las neuropatías hereditarias se dividen en tres categorías principales de acuerdo a sus características clínica: (1) neuropatía hereditaria motora y sensorial, (2) neuropatía hereditaria sensorial y autonómica, y (3) neuropatía hereditaria motora (también llamada atrofia muscular espinal) [ver tabla 4]. En la actualidad se dispone de pruebas genéticas para algunas neuropatías hereditarias (en especial para los defectos en el gen PMP22 en la enfermedad de Charcot-Marie-Tooth de tipo 1). Aunque no existe tratamiento específico para las neuropatías hereditarias, el diagnóstico correcto sigue siendo importante para el pronóstico, educación y consejo genético. En general, las deficiencias en las neuropatías hereditarias progresan en forma muy lenta, la expectancia de vida es normal y la mayoría de los pacientes podrán caminar hasta el final de su vida.
Polineuropatías inmune-inflamatorias
Síndrome de Guillain-Barré El síndrome de Guillain-Barré (SGB), también conocido como polirradiculoneuropatía desmielinizante inflamatoria aguda, es la causa más común de parálisis generalizada aguda en el mundo occidental. La mayoría de los hospitales generales atienden varios pacientes con SGB cada año y el tratamiento suele recaer en médicos generales. La historia natural del SGC casi siempre es favorable, pero la buena evolución depende del cuidado médico y de enfermería durante la mayor deficiencia neurológica.
El diagnóstico de SGB no es difícil una vez que los signos son floridos. Sin embargo, en los primeros días los síntomas y signos pueden ser vagos. Es común que un paciente sea dado de alta del servicio de urgencia con diagnóstico de ansiedad, solo para regresar uno o dos días después con debilidad obvia y progresiva de las extremidades. Lo más frecuente es que el primer síntoma consista en piquetes que comienzan en los pies y se diseminan en forma proximal en cuestión de horas. La debilidad se nota algunas horas a días después. Algunos pacientes solo tienen síntomas motores y no sensitivos. Clásicamente los síntomas comienzan en forma simétrica en la porción distal de las extremidades y progresan en forma proximal (la llamada parálisis ascendente), pero existen variaciones a este patrón, como asimetría y afección predominante de nervios craneales o autonómica. Muchos pacientes sufren dolor profundo en la espalda o extremidades durante las primeras semanas.
La disfunción autonómica puede ser un problema importante en algunos pacientes con SGB, en especial en los que tienen debilidad severa. La fluctuación importante en la presión arterial y la hipotensión refractaria son problemas serios, y puede ocurrir hipertermia, parálisis pupilar y arritmias cardiacas.
La debilidad muscular aumenta durante los primeros días y después permanece estable por días a semanas. Puede variar de leve (v.gr., ligera debilidad a la dorsiflexión) a severa, y ocurre cuadriparesia flácida con parálisis de los músculos respiratorios en hasta el 30 porciento de los pacientes. Es imposible predecir en los primeros 2 días qué tanta debilidad desarrollará cada paciente, por lo tanto, es aconsejable observarlo en el hospital hasta que la severidad sea aparente. Como regla general, la debilidad alcanza su máximo en 14 días. El periodo de debilidad estable antes de que inicie la recuperación dura días a meses, con una duración promedio de 4 semanas. Existe correlación burda entre la severidad de la debilidad y el intervalo antes del inicio de la recuperación. Una vez que comienza la recuperación el paciente suele tener progresos importantes cada semana. Un año después del inicio la mayoría de los pacientes tiene recuperación total o importante, pero hasta el 15 porciento queda confinado a la cama o en silla de ruedas. En alrededor del 3 porciento de los pacientes ocurren episodios recurrentes de SGB, en ocasiones en un lapso de muchos años.
El evento patológico fundamental en el SGB es la destrucción de la mielina de los axones por los macrófagos, que ocurre en parches en el SNP. Se supone que se activa una cascada de eventos que incluyen mecanismos inmunológicos celulares y humorales y con frecuencia se encuentran infiltrados inflamatorios de linfocitos en los nervios examinados por biopsia o posmortem. Se desconoce el desencadenante y las moléculas blanco específicas del ataque inmunológico. En los años recientes ha existido interés sobre el papel potencial de Campylobacter jejuni porque la infección con varios serotipos poco comunes de C. jejuni precede a alrededor del 40 porciento de los casos de SGB.19
El diagnóstico diferencial de la polineuropatía sensorimotora aguda es breve [ver tabla 2]. Antes del desarrollo de la vacuna de la poliomielitis el principal diagnóstico diferencial era entre SGB y poliomielitis, pero esta última es rara en la actualidad. Si existen signos de enfermedad sistémica asociados con la polineuropatía aguda debe pensarse en neuropatía vasculítica, infiltración linfomatosa de las raíces nerviosas, porfiria intermitente aguda, difteria e intoxicación por arsénico. La lesión isquémica de la protuberancia puede producir al inicio cuadriparesia flácida aguda y simular un SGB antes de que se desarrollen los signos de motoneurona superior. En las variantes de SGB que afectan a los nervios craneales (v.gr., síndrome de Miller Fisher) deben excluirse miastenia gravis y botulismo.
El SGB se diagnostica principalmente por sus características clínicas, pero las pruebas de laboratorio pueden apoyar el diagnóstico y excluir otras causas de polineuropatía aguda. El líquido cefalorraquídeo en el SGC característicamente muestra niveles aumentados de proteínas, niveles de glucosa normal y ausencia de pleocitosis. Si se encuentra pleocitosis de mononucleares debe considerarse seriamente infiltración meníngea por un linfoma o carcinoma. Puede ocurrir una polirradiculopatía aguda semejante al SGB pero con pleocitosis en el LCR en el momento de la seroconversión del SIDA [ver adelante, Polineuropatías causadas por enfermedades infecciosas]. Los estudios de conducción nerviosa en el SGB suelen demostrar retraso difuso en la velocidad de conducción o, con más frecuencia, bloqueo proximal de la conducción, y pueden proporcionar cierta información pronóstica. Ni las alteraciones del LCR ni de conducción nerviosa son diagnósticas del SGB, y ambas pueden ser normales, en especial durante los primeros días de la enfermedad.
Una vez que se hace el diagnóstico de SGB la prioridad consiste en vigilar la función de los músculos respiratorios de forma estrecha y estar preparado para intervenir si se desarrolla falla ventilatoria. Es importante la medición frecuente de la capacidad vital, del esfuerzo inspiratorio máximo o de ambos y la evaluación clínica en busca de signos de fatiga de los músculos respiratorios. Un paciente con una capacidad vital de menos de 20 ml/kg puede desarrollar falla ventilatoria franca y debe considerarse la intubación electiva.
Varios estudios clínicos extensos han demostrado que dos tratamientos (plasmaféresis e inmunoglobulina intravenosa [Ig IV]) mejoran el porcentaje de recuperación en pacientes con SGB severo a moderado (v.gr., pacientes incapaces de caminar).20-22 Ambos tratamientos parecen modular el proceso inflamatorio que produce la lesión nerviosa, pero no promueven la remielinización o la regeneración nerviosa. Puede demostrarse un efecto benéfico solo si la plasmaféresis o la Ig IV se administran en las primeras 2 semanas de iniciados los síntomas. Probablemente sea correcto iniciar tratamiento en cuanto se realice el diagnóstico si la debilidad es considerable o si los signos progresan mientras el paciente está en observación. Por lo general se administran 5 fases de tratamiento durante 5 a 10 días, algunos pacientes pueden recaer y requerir mayor tratamiento (v.gr., dos veces por semana durante 3 semanas). El tratamiento combinado (plasmaféresis seguida de IgIV) no ofrece ventajas claras sobre cualquiera de éstos aislado,22 y los esteroides no demostraron ser benéficos cuando se agregaron al tratamiento con plasmaféresis en un estudio controlado extenso.23 Ya que la plasmaféresis y la Ig IV parecen tener la misma eficacia, costos y tasas de recurrencia, muchos clínicos consideran que la Ig IV es el tratamiento de elección porque es más fácil de administrar que la plasmaféresis. Sin embargo, persisten algunas preocupaciones sobre el riesgo de infección por un producto elaborado partir de sangre almacenada, así como incertidumbre sobre si el suplemento de Ig IV es suficiente para satisfacer la demanda.
Debido a que muchos pacientes quedan confinados a la cama durante meses, es indispensable la atención cuidadosa de enfermería. Se requieren medidas para prevenir úlceras de decúbito y trombosis venosas, y el paciente también está en peligro constante de sufrir infecciones urinarias y pulmonares. El dolor es un problema importante en algunos pacientes y parece relacionarse en parte con la inmovilización y en parte con la inflamación y disfunción neurológica. Son benéficos los ejercicios regulares de fisioterapia para mantener rangos de movimiento y el uso juicioso de analgésicos y agentes como la amitriptilina. Un reto especial para todos los miembros del personal es el apoyo psicológico al paciente; existen varios escritos elocuentes sobre la perspectiva del paciente en el SGB.24
Polirradiculoneuropatía desmielinizante inflamatoria crónica Como el SGB, la polirradiculoneuropatía desmielinizante inflamatoria crónica (PDIC) es una neuropatía mediada por un proceso inmune. El evento fisiológico fundamental es la separación de la mielina de los axones por macrófagos, lo que retrasa o bloquea la conducción del impulso nervioso, causando debilidad y pérdida sensorial. Desde el punto de vista clínico, la PDIC difiere del SGB en varios aspectos importantes. El inicio de la PDIC es insidioso, con síntomas y signos que se desarrollan en semanas a meses, comparado con el curso inicial rápido del SGB. A diferencia del SGB, con evolución monofásica y autolimitada, la historia natural de la PDIC es variable. El patrón habitual consiste en progresión lenta en meses, produciendo incapacidad moderada crónica. Algunos pacientes tienen un curso de recidivas y remisiones y muy pocos presentan remisión espontánea gradual. La afección autonómica o respiratoria importante, que es común en el SGB, es poco habitual en la PDIC.
Ningún dato clínico o de laboratorio es patognomónico de PDIC y el diagnóstico se hace por la combinación de la historia clínica, signos de neuropatía, evidencia electromiográfica de desmielinización y LCR acelular con aumento en el nivel de proteínas. Puede ocurrir un tipo semejante de neuropatía crónica en pacientes con gamopatía monoclonal, mieloma osteoesclerótico e infección por VIH (ver adelante).
La eficacia de diversos tratamientos para la PDIC se ha demostrado en estudios controlados. La plasmaféresis25 y la Ig IV26 producen mejoría en 2 a 3 semanas en la mayoría de los pacientes, pero ésta es breve y los pacientes requieren de tratamientos repetidos o alternativos por tiempo prolongado. La prednisona en dosis altas también es eficaz,27 pero la respuesta suele ser menos impresionante y más lenta y el paciente enfrenta las complicaciones potenciales asociadas con el uso prolongado de esteroides. Existen reportes anecdóticos sobre el uso de fármacos inmunomoduladores en el tratamiento de la PDIC, incluyendo ciclosporina, azatioprina, interferón-alfa y ciclofosfamida. Son importantes la vigilancia cuidadosa del daño neurológico y el tratamiento por neurólogos con experiencia en PDIC.
Polineuropatía asociada a proteína monoclonal La polineuropatía y la gamopatía monoclonal pueden ocurrir en algunos pacientes que tienen amiloidosis, mieloma múltiple, mieloma osteoesclerótico, macroglobulinemia de Waldenström o linfoma. Si no se encuentra ninguna de estas alteraciones hematológicas debe pensarse que el paciente tiene gamopatía monoclonal de significado no determinado (GMSN). La relación entre la GMSN y la neuropatía no está bien establecida, pero evidencias epidemiológicas y patológicas sugieren que la GMSN causa la neuropatía.
La neuropatía por GMSN tiene características clínicas y electrofisiológicas variables. Suele existir evidencia electrofisiológica y patológica de desmielinización prominente y el cuadro clínico es muy semejante al de la PDIC. Algunos pacientes tienen neuropatía axonal dolorosa con deficiencias leves. La neuropatía asociada con IgM tiende a ser más severa, con más ataxia y más evidencia de retraso y dispersión en la conducción nerviosa. Esto indica que existe cierta relación entre la clase de inmunoglobulina y las características de la neuropatía.28
La neuropatía de la GMSN suele ser de progresión lenta, aunque el grado de incapacidad es variable. Los enfoques terapéuticos se dirigen a suprimir la producción de proteína monoclonal o a eliminarla de la circulación. Existen reportes anecdóticos de respuesta al clorambucil, melfalán y prednisona. Un estudio controlado mostró que la plasmaféresis es benéfica en pacientes con gamopatías de IgG e IgA pero no en las de IgM.29
Neuropatía por amiloide La polineuropatía caracterizada por depósito de amiloide en los nervios ocurre en dos condiciones: como manifestación en alrededor del 15 porciento de los pacientes con amiloidosis sistémica, en los que la proteína amiloidogénica es una inmunoglobulina, y como un trastorno autosómico dominante, la polineuropatía familiar por amiloide (PFA), en la que el amiloide consiste en formas mutadas de transtirretina, una proteína sérica normal.30 Ambos tipos consisten en polineuropatía simétrica sensorimotora que se diferencía por la presencia de características autonómicas importantes. La amiloidosis también es causa poco frecuente de síndrome de tunel del carpo.
El diagnóstico de neuropatía por amiloide se realiza por biopsia de nervio. La neuropatía adquirida y la hereditaria son histopatológicamente semejantes pero pueden distinguirse por análisis inmunohistoquímico. Los estudios de tratamiento con melfalán, esteroides y colchicina han tenido poco impacto sobre la evolución de los pacientes con amiloidosis primaria, incluyendo mejoría de la neuropatía. A diferencia de la amiloidosis primaria, la neuropatía suele ser la causa principal de incapacidad en la PFA y la mayoría de los pacientes sobreviven durante una década o más después del diagnóstico. El trasplante hepático detiene la progresión de la neuropatía en la PFA.31
Mieloma osteoesclerótico El mieloma osteoesclerótico es una variante de mieloma en el que los pacientes tienen una o en ocasiones varias lesiones óseas osteoescleróticas. La biopsia revela proliferación maligna de células plasmáticas. Con frecuencia estas lesiones ocurren dentro de una constelación que incluye polineuropatía, organomegalia, alteraciones endócrinas, gamopatía monoclonal y cambios en la piel (síndrome de POEMS). La polineuropatía típicamente es desmielinizante y recuerda la de la PDIC. Es importante detectar el síndrome de POEMS porque puede tener importante mejoría clínica con la radiación de la lesión ósea.
Neuropatía paraneoplásica Se han descrito desde 1950 varios síndomes neurológicos no metastásicos característicos que se desarrollan en pacientes con cáncer. Se piensa que los síndromes paraneoplásicos tienen una base inmunológica, desarrollándose como consecuencia del intento del huésped para montar una respuesta inmune contra la neoplasia32.
Neuropatía por vasculitis. La neuropatía es una manifestación frecuente de ciertas vasculitis sistémicas. La neuropatía por vasculitis es isquémica, consecuencia de afección de los vasos de nutrición del nervio por el proceso inflamatorio. Debido a la abundante irrigación sanguínea del nervio y su resistencia relativa a la lesión isquémica, el desarrollo de neuropatía en la vasculitis implica afección extensa de los vasos. La vasculitis tiende a ser en parches y es común la asimetría en la afección nerviosa, de modo que se afectan nervios individuales y se respetan los vecinos; este es el síndrome clásico de mononeuropatía múltiple. Sin embargo, al avanzar la afección vascular y afectarse más nervios puede ser difícil identificar el patrón de mononeuropatía múltiple. Alrededor del 30 porciento de los casos con neuropatía por vasculitis tienen polineuropatías simétricas en el momento del diagnóstico inicial, 30 porciento polineuropatías asimétricas y 40 porciento mononeuropatías múltiples.33
Entre los pacientes con neuropatía por vasculitis, las vasculitis sistémicas más frecuentes son la poliarteritis nodosa, la vasculitis reumatoide, el síndrome de Sjögren, la granulomatosis de Wegener y la angeitis granulomatosa alérgica (síndrome de Churg-Strauss). La neuropatía es especialmente común en la poliarteritis nodosa, ocurriendo en por lo menos la mitad de los casos. Las características clínicas y neuropatológicas de la neuropatía en estos trastornos son semejantes y el diagnóstico específico depende de las características sistémicas y no de las neurológicas. Puede sospecharse neuropatía por vasculitis por el cuadro clínico, pero el diagnóstico definitivo depende de la biopsia de nervio. Con frecuencia se encuentran anticuerpos anticitoplasma de neutrófilo en la neuropatía por vasculitis, pero las pruebas falsas positivas limitan su utilidad diagnóstica.34
La neuropatía por vasculitis se maneja tratando la enfermedad subyacente. Esto generalmente requiere de dosis altas de prednisona, ciclofosfamida o ambas. En la vasculitis la neuropatía rara vez es la causa de la muerte, aunque puede producir incapacidad significativa. Si pueden tratarse con éxito los eventos graves, como la falla renal o cardiorrespiratoria, el prospecto de mejoría neurológica es bueno aunque la mejoría puede tardar muchos meses.35
Los pacientes con manifestaciones clínicas y neuropatológicas de neuropatía vasculítica en ocasiones no tienen evidencia de vasculitis sistémica. Este síndrome, denominado neuropatía vasculítica no sistémica, parece tener una historia natural relativamente benigna y puede ser difícil justificar el tratamiento inmunosupresor por los riesgos asociados.36
Neuropatías relacionadas con otras enfermedades del tejido conjuntivo La neuropatía periférica puede ocurrir en otras enfermedades del tejido conjuntivo, como el lupus eritematoso generalizado, aunque puede ser difícil asegurar si la neuropatía es una complicación directa del padecimiento o un efecto secundario de otra complicación (v.gr., secundaria a insuficiencia renal). Suele suponerse que la neuropatía en estos casos tiene un fondo inflamatorio, pero existen pocos estudios patológicos que apoyen esta creencia. Se ha descrito en los pacientes con síndrome de Sjögren una neuronopatía sensorial atáxica semejante a la del síndrome paraneoplásico, que es causada por infiltración inflamatoria de las raíces dorsales de los ganglios.37 También ocurre neuropatía sensorial inflamatoria en los pacientes con síndrome sicca que no tienen características extraglandulares de síndrome de Sjögren.38
Neuropatías causadas por toxinas y deficiencias nutricionales
Las sustancias tóxicas para los nervios periféricos incluyen diversos químicos industriales, compuestos naturales y fármacos. La mayoría de las neuropatías tóxicas comienzan en forma distal, progresan de modo insidioso durante semanas a meses y tienen características electrofisiológicas de neuropatía axonal. Con algunas excepeciones, la degeneración axonal inespecífica es la principal característica histológica y la biopsia de nervio rara vez es útil para hacer el diagnóstico.
Fármacos La neuropatía inducida por fármacos es un problema común, en especial en el ambiente hospitalario. La historia cuidadosa de los medicamentos recibidos es parte importante de la investigación de cualquier polineuropatía. La neuropatía periférica es un efecto adverso bien establecido de muchos fármacos [ver tabla 5]. En circunstancias ideales, antes de que un medicamento se etiquete como potencialmente neurotóxico, deben haberse descartado en el paciente afectado otras causas potenciales de neuropatía y la suspensión del medicamento debe causar cierta mejoría clínica. Es importante saber que la recuperación puede tardar muchos meses. En algunas neuropatías tóxicas el paciente presenta un fenómeno conocido como persistencia, en el que la neuropatía continúa empeorando por algunas semanas después de que cesa la exposición. Para la mayoría de las neurotoxinas bien establecidas existe evidencia experimental de toxicidad en animales y cultivos de células neuronales. Con algunos medicamentos antineoplásicos (v.gr., cisplatino) la neuropatía puede ser el efecto adverso que limita la dosis. En estudios en animales la neuropatía por cisplatino puede prevenirse o aliviarse por la coadministración de neurotrofinas, como el NT-3, una estrategia que quizá podrá aplicarse a humanos.39
Químicos industriales La historia laboral del paciente puede ser importante porque diversos químicos industriales son neurotóxicos [ver tabla 6]. La mayoría de estos compuestos fueron detectados como neurotóxicos después de acúmulos de casos que aparecieron en trabajadores de industrias específicas y el conocimiento de este riesgo ha reducido la incidencia de casos. La mayoría de estos químicos producen neuropatía axonal con cambios patológicos inespecíficos. Demostrar que una neuropatía es causada por la exposición a un químico requiere de trabajo epidemiológico cuidadoso apoyado por estudios en animales o cultivos de tejidos.
|
||
* La exposición a estos compuestos también se observa en la inhalación de pegamento y otros abusos de solventes |
Metales Además de ser causada por medicamentos que contienen oro y platino, puede ocurrir neuropatía por intoxicación con otros metales. La exposición con frecuencia es resultado de intentos homicidas o suicidas, de modo que la historia tiene valor dudoso. El diagnóstico puede hacerse a partir de las manifestaciones clínicas asociadas. Por ejemplo, la neuropatía por plomo es principalmente motora, con predilección por las extremidades superiores, y se asocia con dolor abdominal, constipación y anemia. Además de causar neuropatía, la intoxicación por arsénico produce dolor abdominal, vómito, diarrea, cambios en la piel y uñas y pancitopenia. La neuropatía por talio se distingue por alopecia y dolor abdominal. Los compuestos de mercurio orgánicos e inorgánicos pueden producir neuropatía, aunque predominan los efectos sobre el SNC .
El diagnóstico de neuropatía por metales se basa en la demostración de mayor excreción urinaria del metal o mayores niveles en el cabello o las uñas. Debe tenerse la sospecha clínica para solicitar estos estudios, que no son parte de la investigación de rutina de una polineuropatía.
Etanol Es común la neuropatía distal, con frecuencia dolorosa, en los alcohólicos crónicos. Los principales síntomas son quemadura, dolor lancinante y parestesias en los pies y en ocasiones en las manos. La pérdida sensorial o la hipersensibilidad dolorosa en los pies, la pérdida de reflejos aquíleos y la debilidad distal leve forman el cuadro clínico típico. No se conoce si la neuropatía es causada por un efecto tóxico directo del etanol, por desnutrición o por ambos.
Los intentos por producir neuropatía con etanol en animales bien alimentados no han tenido éxito, aunque en cultivos de células neuronales puede producirse inhibición del crecimiento con concentraciones moderadamente altas de etanol. Un estudio danés examinó en forma cuidadosa una cohorte de bebedores de cerveza alcohólicos con neuropatía y no encontró diferencias clínicas, electrofisiológicas o histológicas entre los que estaban bien nutridos y los desnutridos.40 La cerveza danesa está suplementada con tiamina y piridoxina, de modo que es poco probable que la deficiencia de estas vitaminas sea la causa de neuropatía en estos pacientes.
Aunque la patogenia de la neuropatía en alcohólicas es motivo de controversia, el cuadro clínico es característico porque generalmente existen otros estigmas de alcoholismo, incluyendo hepatopatía crónica, deterioro de la memoria y ataxia de la marcha. Los cálculos sobre la ingesta de alcohol son muy poco confiables, pero en el estudio danés se desarrolló neuropatía solo en los pacientes que consumieron 3 L de cerveza o 300 ml de bebidas alcohólicas de alta graduación diario por lo menos durante 3 años. Un corolario importante a estas observaciones es que un paciente con neuropatía que bebe en forma moderada, está bien nutrido y no tiene datos de enfermedad hepática crónica es poco probable que tenga neuropatía alcohólica, y deben investigarse otras causas de la misma.
El tratamiento de la neuropatía alcohólica es simple en principio pero difícil de aplicar. Debe suspenderse la ingestión de etanol y asegurar una nutrición adecuada. Si se logran estas medidas puede esperarse mejoría, aunque ésta puede tardar meses y ser incompleta.
Deficiencias nutricionales La polineuropatía puede ser una manifestación del ayuno, como el observado en víctimas de hambrunas o en prisioneros de guerra. Los componentes precisos de la dieta responsables de la neuropatía por ayuno no son claros, aunque una o más vitaminas B parecen ser cruciales. Ciertas deficiencias de vitamina pueden causar neuropatía en circunstancias específicas, pero no existe base fisiológica para la prescripción rutinaria de multivitamínicos para la neuropatía en pacientes con dietas normales.
La deficiencia de tiamina (vitamina B1) produce beriberi, cuyas principales características son polineuropatía y falla cardiaca. La neuropatía es distal y axonal, con síntomas sensoriales dolorosos. Al progresar puede presentarse debilidad axonal. En las primeras descripciones de beriberi se describió afección de los nervios craneales.
La deficiencia de piridoxina (vitamina B6) es responsable de la neuropatía causada por isoniacida, que aumenta la excreción de piridoxina. La administración de piridoxina con la isoniacida evita la neuropatía. Sin embargo, la administración excesiva de piridoxina (que se usó en los años 70) produce una neuronopatía sensorial severa.
La polineuropatía leve puede ser parte del síndrome neurológico producido por deficiencia de cobalamina (vitamina B12). Sin embargo, es posible que la deficiencia de esta vitamina no se presente solo como polineuropatía y las principales manifestaciones clínicas son resultado de mielopatía (degeneración combinada subaguda).
La deficiencia de vitamina E por malabsorción puede deberse a un síndrome atáxico causado por degeneración de los procesos periféricos y centrales de las neuronas en los ganglios de las raíces dorsales. Puede existir afección cerebelosa en algunos pacientes.
Polineuropatías causadas por enfermedades infecciosas
Lepra La lepra, una infección micobacteriana de los nervios periféricos, parece ser la causa más común de polineuropatía en el mundo. La mayoría de los casos ocurren en regiones tropicales y subtropicales, aunque existen focos endémicos a lo largo de la costa del Golfo de Florida y Louisiana. La mayoría de los pacientes con lepra en Norteamérica y Europa son inmigrantes de países en donde la lepra es común.
La pérdida sensorial es el síntoma cardinal de la lepra. Con frecuencia se descubre por una lesión no dolorosa. Debido a los requerimientos de temperatura de Mycobacterium leprae, los nervios sensoriales cutáneos y los nervios mixtos en partes del cuerpo con baja temperatura ambiental son los más afectados. Esto causa una distribución de signos diferente a los de otras polineuropatías, con pérdida sensorial en los pabellones auditivos, los arcos cigomáticos y las superficies extensoras de las articulaciones. Los nervios grandes se afectan en los trayectos en que viajan cercanos a la superficie (v.gr., el nervio cubital a nivel del codo). La afección de los nervios cutáneos suele estar bien delimitada, en especial en la forma tuberculoide de la lepra, y se afectan la dermis y epidermis suprayacentes, produciendo la clásica mancha anestésica. No ocurre debilidad motora hasta que la pérdida sensorial está bien establecida. La debilidad suele ser en parches y asimétrica y puede sugerir otras causas de mononeuropatía múltiple. El diagnóstico de lepra se hace por biopsia de nervio o piel, usando el método de Fite para teñir e identificar a M. leprae.
La lepra es una enfermedad curable. El grado de recuperación en casos avanzados puede ser limitado, de modo que es importante que la enfermedad sea diagnosticada y tratada antes de que se desarrollen deficiencias neuropáticas importantes.
Infección por VIH Ocurren varios trastornos del SNP en los pacientes con infección por VIH, algunos en las fases tempranas de la infección y otros solo después de la progresión a SIDA . Es muy común la neuropatía dolorosa en los pacientes con SIDA. El principal síntoma es una molestia ardorosa continua, principalmente en los pies, en donde existe cierto grado de pérdida sensorial. La afección motora suele ser leve, aunque los pacientes están debilitados por infecciones concomitantes y pérdida de peso. En ocasiones puede identificarse la causa (por ejemplo, deficiencia de vitamina B12 o tratamiento con un agente retroviral conocido por ser neurotóxico como 2,3-dideoxicitidina o zalcitabina)41 pero en algunos pacientes la causa no es clara. Se ha detectado el genoma del VIH en las neuronas de las raíces dorsales y en células satélite en pacientes con SIDA y neuropatía,42 y la expresión del genoma del VIH en ratones transgénicos produce enfermedad del nervio periférico.43 Aún queda por demostrarse si la neuropatía en humanos con VIH es causada por infección directa del nervio por el virus.
Típicamente ocurren SGB y PDIC en la fase temprana de la infección por VIH. Estas pueden ser las manifestaciones iniciales de la infección y debe considerarse realizar una prueba de VIH en los pacientes con estos tipos de neuropatía. El cuadro neurológico y la respuesta al tratamiento son semejantes a los de los pacientes sin infección por VIH, excepto porque suele existir pleocitosis linfocítica en el LCR.
Puede ocurrir un síndrome de mononeuropatía múltiple en los pacientes VIH positivos. La biopsia de nervio puede mostrar infiltrados inflamatorios perivasculares, vasculitis necrosante o inclusiones por citomegalovirus (CMV). Los pacientes con evidencia de infección por CMV pueden responder a gangiclovir44. En una serie de pacientes se ha reportado la presencia de un síndrome de linfocitosis infiltrativa difusa y neuropatía axonal, las biopsias de nervio mostraron infiltrados perivasculares de linfocitos CD8+ y la neuropatía cedió con esteroides o zidovudina.45
La polirradiculopatía lumbosacra aguda es un síndrome devastador de debilidad en miembros inferiores y pérdida sensorial perineal, desarrollándose retención urinaria en 1 a 2 semanas, por lo general en pacientes con SIDA avanzado. El LCR muestra pleocitosis polimorfonuclear característica. En la mayoría de los casos existe evidencia de infección concomitante por CMV y el cuadro neurológico es causado por invasión de las raíces nerviosas lumbosacras por el CMV. El diagnóstico pronto y el tratamiento con ganciclovir pueden producir mejoría. El linfoma puede producir un cuadro clínico semejante.46
Enfermedad de Lyme La enfermedad de Lyme es causada por la espiroqueta Borrelia burgdorferi, que se trasmite a los humanos por garrapatas ixodes. La enfermedad ocurre en todo el mundo pero es más común en el noreste de los Estados Unidos y el norte de Europa. La neuropatía periférica puede ocurrir en la fase temprana o tardía de la enfermedad de Lyme. No es claro si las manifestaciones neurológicas son causadas en forma directa por la invasión de espiroquetas en el tejido nervioso o por la respuesta inmunológica del huésped al organismo.
Las principales manifestaciones neurológicas tempranas son neropatías craneales, radiculopatías espinales o ambas. La cefalea y la rigidez de cuello pueden acompañar a los síntomas de los nervios perifércos, reflejando inflamación meníngea. La neuropatía facial es la neuropatía craneal más común y con frecuencia se diagnostica en forma equívoca como parálisis de Bell. Sin embargo, a diferencia de la parálisis de Bell idiopática, la neuropatía facial bilateral es común en la enfermedad de Lyme. Ocurren otras neuropatías craneales con menos frecuencia.
La afección de las raíces espinales comienza en forma típica con dolor en una distribución radicular, seguido de debilidad en 1 a 4 semanas. La debilidad suele ser asimétrica y en parches, recordando una mononeuropatía múltiple. Además de los signos radiculares dominantes, existe con frecuencia evidencia electrofisiológica de polineuropatía diseminada leve. Tanto la neuropatía craneal como espinal de la fase temprana de la enfermedad de Lyme tienen una historia natural favorable, con recuperación en semanas a meses. El tratamiento con antibióticos puede acelerar la recuperación.
En la enfermedad de Lyme tardía los pacientes pueden desarrollar polineuropatía distal leve o dolor radicular con signos sensoriales.47 Muchos de estos pacientes tendrán sintomas previos de enfermedad localizada o diseminada en fase temprana. A diferencia de la neuropatía craneal y la radiculoneuropatía de la fase temprana, la neuropatía periférica de la fase terdía no se resuelve con el tratamiento.
El diagnóstico de neuropatía de Lyme se basa en la historia de posible exposición a la garrapata, síntomas y signos compatibles y pruebas serológicas positivas para B. burgdorferi. En la enfermedad en fase temprana la afección neurológica se acompaña casi siempre de pleocitosis linfocítica en el LCR, pero en la fase tardía la pleocitosis es excepcional y el LCR puede ser normal. La biopsia del nervio sural en la enfermedad en fase temprana o tardía muestra degeneración axonal e infiltrados inflamatorios perivasculares, pero estos datos son inespecíficos. En especial en la enfermedad en fase tardía la respuesta a un esquema de antibióticos puede proporcionar la evidencia más contundente del diagnóstico.
Infección por virus varicela-zoster El virus varicela-zoster (VVZ), miembro de la familia de los herpesvirus humanos, es el patógeno viral más común para el SNP.
El contacto inicial con el VVZ, por lo general en la infancia, causa viremia transitoria seguida de una erupción vesicular difusa de la piel y mucosas (varicela). Si la respuesta inmune es apropiada el virus es controlado y las lesiones vesiculares sanan. Algunos viriones pueden entrar en los axones sensoriales cutáneos y son transportados en forma retrógrada a los cuerpos neuronales sensoriales en las raíces dorsales o los ganglios craneales, en donde permanecen en forma inactiva.
Años después el virus puede regresar a un estado activo y proliferativo por alteraciones en la función inmune que aún no se comprenden del todo. Los efectos citopáticos de la replicación del VVZ y la respuesta inmune que se despierta producen una ganglionitis necrótica inflamatoria intensa que ocasiona alteración en la sensibilidad de un dermatoma y la erupción cutánea vesicular en el dermatoma conocida como herpes zoster.
El herpes zoster es más común en personas mayores de 60 años, aunque también ocurre en jóvenes. La incidencia es mucho mayor en pacientes inmunosuprimidos, en los que existe también riesgo de infección diseminada por VVZ, que tiene una mortalidad considerable. El herpes zoster suele ser unilateral, afectando uno a tres dermatomas adyacentes. Los dermatomas torácicos y del trigémino (sobre todo el oftálmico) son los más afectados.
El síndroma inicial es el dolor en el área del dermatoma, seguido en 3 a 7 días por la erupción vesicular. Es difícil demostrar la pérdida sensorial sino hasta que las lesiones cutáneas comienzan a sanar. Se reporta debilidad motora en hasta el 30 porciento de los pacientes, que puede no notarse por el dolor existente. Las lesiones cutáneas persisten por 7 a 10 días y después se resuelven, dejando con frecuencia áreas de menor pigmentación y cicatrización. Es de esperar que la debilidad motora mejore en forma espontánea en la mayoría de los pacientes. El dolor también mejora con lentitud, excepto en los pacientes que desarrollan neuralgia posherpética (NPH).
La NPH suele definirse como el dolor que persiste por más de 4 a 8 semanas después de que sanan las lesiones cutáneas. El riesgo de desarrollar NPH después de un episodio de herpes zoster aumenta con la edad: hasta 45 porciento en los pacientes mayores de 65 años. Los pacientes con NPH sufren dolor ardoroso continuo en el dermatoma, al que se agregan dolores lancinantes breves. Con frecuencia existe también hiperpatía en el dermatoma afectado, por lo que el tacto de la ropa o incluso el cabello pueden causar dolor intenso. Algunos pacientes con NPH presentan mejoría gradual en semanas a meses, pero no es raro que los síntomas persistan por años. La prevención de la NPH es la justificación más importante para usar agentes antivirales en el paciente inmunocompetente. Un estudio controlado con placebo y extenso de famciclovir mostró reducción clara en la incidencia de NPH, en especial en los ancianos.48 Se supone que el aciclovir ofrece un beneficio semejante, aunque esto no se ha demostrado. En un estudio, la adición de prednisolona a los esquemas de 7 o 21 días de aciclovir no alteró la frecuencia de NPH,49 a pesar de que los esteroides pueden disminuir el dolor en la fase aguda.
La NPH implica un reto terapéutico formidable. La amitriptilina u otros medicamentos tricíclicos son los agentes de primera línea y ayudan en cierto grado a la mayoría de los pacientes. El dolor lancinante puede disminuirse con carbamacepina u otros anticonvulsivantes. Algunos clínicos sugieren la aplicación tópica de anestésicos locales y el uso por tiempo prolongado de opioides.50
Tratamiento de los síntomas de neuropatía
Los síntomas de neuropatía pueden ser tratables incluso si la causa de la neuropatía no lo es o se desconoce. Con medidas simples muchos pacientes pueden tener alivio importante de los síntomas.
La ortosis para pie-tobillo es un aditamiento simple que compensa el pie caído. Su principal beneficio es brindar mayor estabilidad al tobillo, causando mejor balance y evitando la tendencia de que los ortejos se atoren en los bordes, irregularidades y alfombras. Las ortosis ligeras, bien adecuadas y que se colocan dentro del zapato suelen ser las más cómodas. Los pacientes con debilidad de la muñeca y los extensores pueden ser ayudados por una férula que mantenga la muñeca y dedos en posición neutra. Por desgracia este tipo de férula es de poca ayuda si existe debilidad concomitante de los músculos intrínsecos de la mano.
El dolor, en especial en los pies, suele acompañarse de alteraciones sensoriales de neuropatía. Paradójicamente, en las polineuropatías con dolor prominente las deficiencias neurológicas suelen ser leves y existe disociación entre la incapacidad y el nivel de molestia para el paciente. El paciente puede interpretar el dolor como un signo de un padecimiento serio que pone en peligro su independencia o su vida. En ocasiones brindar confianza y explicar al paciente la poca función neurológica que se ha perdido ayuda a que éste se adapte mejor al dolor neuropático.
Las medidas no farmacológicas pueden ser tan eficaces como los medicamentos. Debe ponerse atención cuidadosa al calzado. Se aconseja usar zapatos amplios, de suela suave y calcetín grueso. El dolor neuropático suele agravarse con las temperaturas extremas (especialmente con el calor) y las sandalias abiertas pueden causar alivio. El peso prolongado suele empeorar el dolor neuropático, los pacientes que requieren estar de pie en su trabajo mejoran con periodos breves y frecuentes de descanso sentados.
El mojar los pies produce alivio breve en muchos pacientes. El meter los pies hasta el tobillo en agua de la llave fría (sin hielo) durante 15 a 20 minutos puede ser muy útil en el momento de acostarse. Aunque el alivio es breve, puede ser suficiente para permitir que el paciente se duerma con tranquilidad. Para algunos pacientes el calor es mejor que el frío, y otros encuentran que alternar frío y calor (los baños llamados de contraste) proporciona mayor alivio. Es conveniente la inspección diaria de los pies por posibles lesiones (un hábito importante que los pacientes con neuropatía deben desarrollar), así como el uso de calcetines por la noche.
Los medicamentos pueden ser útiles en el manejo del dolor neuropático, aunque los objetivos del tratamiento deben ser realistas. Es poco probable que el dolor se alivie por completo. El objetivo debe ser hacer el dolor más tolerable sin aumentar efectos adversos importantes por el medicamento. De los muchos fármacos empleados para el dolor neuropático la amitriptilina y la carbamacepina son los más usados. Los paroxismos de dolor lancinante responden más a carbamacepina (200 mg al día), mientras que la amitriptilina (10 a 30 mg a la hora de acostarse) suele ser de elección para el adormedimiento ardoroso continuo. Debe intentarse un periodo de prueba de por lo menos un mes antes de tomar conclusiones sobre la utilidad del medicamento. Además de la amitriptilina y la carbamacepina, los salicilatos y otros analgésicos simples causan alivio en algunos pacientes. Como en el caso de otros tipos de dolor crónico, es mejor evitar los narcóticos.
Si los esquemas adecuados de amitriptilina o carbamacepina no son útiles, pueden intentarse varios fármacos de segunda elección, incluyendo baclofén, mexiletina y prazosín. El ungüento de capsaicina tópica causa depleción del neurotrasmisor sustancia P en el asta dorsal de la médula espinal y ayuda en algunos pacientes. Sin embargo, para la mayoría de los pacientes el costo y las molestias de la capsaicina tópica superan cualquier beneficio. El gabapentin, uno de los nuevos anticonvulsivantes, se usa cada vez más para el dolor neuropático, aunque las evidencias de su eficacia hasta la fecha son principalmente anecdóticas.51
Algunos pacientes no mejoran con ninguna de estas medidas y representan problemas de manejo. La depresión concomitante puede complicar la situación y en estos casos será útil referir al paciente al psiquiatra. También debe considerarse enviar al paciente a una clínica multidisciplinaria de dolor.
Figura 1 George Kelvin.
Bibliografía
-
Ide C: Peripheral nerve regeneration. Neurosci Res 25:101,
1996
-
Murakami S, Mizobuchi M, Nakashiro Y, et al: Bell palsy and
herpes simplex virus: identification of viral DNA in endoneurial fluid
and muscle. Ann Intern Med 124:27, 1996
-
Adour KK, Ruboyiane J, Van Doersten PG, et al: Bell's palsy
treatment with acyclovir and prednisone compared with prednisone alone:
a double-blind, randomized, controlled trial. Ann Otol Rhinol Laryngol
105:371, 1996 [PMID
8651631]
-
Suarez GA, Giannini C, Bosch EP, et al: Immune brachial plexus
neuropathy: suggestive evidence for an inflammatory-immune pathogenesis.
Neurology 46:559, 1996 [PMID
8614534]
-
Said G, Goulon-Goeau C, Lacroix C, et al: Nerve biopsy findings
in different patterns of proximal diabetic neuropathy. Ann Neurol 35:559,
1994 [PMID
8179302]
-
Dyck PJ, Oviatt KF, Lambert EH: Intensive evaluation of referred
unclassified neuropathies yields improved diagnosis. Ann Neurol 10:222,
1981 [PMID
7294727]
-
Dyck PJ, Kratz KM, Karnes JL, et al: The prevalence by staged
severity of various types of diabetic neuropathy, retinopathy, and nephropathy
in a population-based cohort: the Rochester Diabetic Neuropathy Study.
Neurology 43:817, 1993 [PMID
8469345]
-
Dyck PJ, Davies JL, Litchy WJ, et al: Longitudinal assessment
of diabetic polyneuropathy using a composite score in the Rochester Diabetic
Neuropathy Study cohort. Neurology 49:229, 1997 [PMID
9222195]
-
Dyck PJ, Zimmerman BR, Vilen TH, et al: Nerve glucose, fructose,
sorbitol, myo-inositol, and fiber degeneration and regeneration
in diabetic neuropathy.
-
Boulton AJM, Levin S, Comstock J: A multicentre trial of
the aldose-reductase inhibitor, tolrestat, in patients with symptomatic
diabetic neuropathy. Diabetologia 33:431, 1990
-
Dyck PJ: Hypoxic neuropathy: does hypoxia play a role in
diabetic neuropathy? Neurology 39:111, 1989
-
Low PA, Tuck RR, Dyck PJ, et al: Prevention of some electrophysiologic
and biochemical abnormalities with oxygen supplementation in experimental
diabetic neuropathy. Proc Natl Acad Sci USA 81:6894, 1984 [PMID
6593734]
-
The effect of intensive diabetes therapy on the development
and progression of neuropathy. The Diabetes Control and Complications Trial
Research Group. Ann Intern Med 122:561, 1995
-
Navarro X, Sutherland DER, Kennedy WR: Long-term effects
of pancreatic transplantation on diabetic neuropathy. Ann Neurol 42:727,
1997 [PMID
9392572]
-
Albers JW, Andersen H, Arezzo JC, et al: Diabetic polyneuropathy
in controlled clinical trials: consensus report of the Peripheral Nerve
Society. Ann Neurol 38:478, 1995
-
Zochodne DW, Bolton CF, Wells GA, et al: Critical illness
polyneuropathy: a complication of sepsis and multiple organ failure. Brain
110:819, 1987 [PMID
3651796]
-
Lacomis D, Giuliani MJ, Van Cott A, et al: Acute myopathy
of intensive care: clinical, electromyographic, and pathological aspects.
Ann Neurol 40:645, 1996 [PMID
8871585]
-
Murakami T, Garcia CA, Reiter LT, et al: Charcot-Marie-Tooth
disease and related inherited neuropathies. Medicine (Baltimore) 75:233,
1996 [PMID
8862346]
-
Rees JH, Soudain SE, Gregson NA, et al: Campylobacter
jejuni infection and Guillain-Barre syndrome. N Engl J Med 333:1374,
1995 [PMID
7477117]
-
Plasmapheresis and acute Guillain-Barre syndrome. The Guillain-Barre
Syndrome Study Group. Neurology 35:1096, 1985
-
A randomized trial comparing intravenous immune globulin
and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study
Group. N Engl J Med 326:1123, 1992
-
Randomised trial of plasma exchange, intravenous immunoglobulin,
and combined treatments in Guillain-Barre syndrome. Plasma Exchange/Sandoglobulin
Guillain-Barre Syndrome Trial Group. Lancet 349:225, 1997
-
Double-blind trial of intravenous methylprednisolone in Guillain-Barre
syndrome. Guillain-Barre Syndrome Steroid Trial Group. Lancet 341:586,
1993
-
Bowes D: The doctor as patient: an encounter with Guillain-Barre
syndrome. Can Med Assoc J 131:1343, 1984
-
Hahn AF, Bolton CF, Pillay N, et al: Plasma exchange therapy
in chronic inflammatory demyelinating polyneuropathy: a double-blind, sham-controlled,
crossover study. Brain 119:1055, 1996 [PMID
8813270]
-
Dyck PJ, Litchy WJ, Kratz KM, et al: A plasma exchange versus
immune globulin infusion trial in chronic inflammatory demyelinating polyradiculoneuropathy.
Ann Neurol 36:838, 1994 [PMID
7998769]
-
Dyck PJ, O'Brien PC, Oviatt KF, et al: Prednisone improves
chronic inflammatory demyelinating polyradiculoneuropathy more than no
treatment. Ann Neurol 11:136, 1982
-
Gosselin S, Kyle RA, Dyck PJ: Neuropathy associated with
monoclonal gammopathies of undetermined significance. Ann Neurol 30:54,
1991 [PMID
1656848]
-
Dyck PJ, Low PA, Windebank AJ, et al: Plasma exchange in
polyneuropathy associated with monoclonal gammopathy of undetermined significance.
N Engl J Med 325:1482, 1991
-
Reilly MM, Adams D, Booth DR, et al: Transthyretin gene analysis
in European patients with suspected familial amyloid polyneuropathy. Brain
118:849, 1995 [PMID
7655883]
-
Bergethon PR, Sabin TD, Lewis D, et al: Improvement in the
polyneuropathy associated with familial amyloid polyneuropathy after liver
transplantation. Neurology 47:944, 1996
-
Chalk CH, Windebank AJ, Kimmel DW, et al: The distinctive
clinical features of paraneoplastic sensory neuronopathy. Can J Neurol
Sci 19:346, 1992 [PMID
1393844]
-
Hawke SHB, Davies L, Pamphlett R, et al: Vasculitic neuropathy:
a clinical and pathological study. Brain 114:2175, 1991
-
Chalk CH, Homburger HA, Dyck PJ: Anti-neutrophil cytoplasmic
antibodies in vasculitic peripheral neuropathy. Neurology 43:1826, 1993
[PMID
8414041]
-
Chang RW, Bell CL, Hallet M: Clinical characteristics and
prognosis in vasculitic mononeuropathy multiplex. Arch Neurol 41:618, 1984
[PMID
6326716]
-
Dyck PJ, Benstead TJ, Conn DL, et al: Nonsystemic vasculitic
neuropathy. Brain 110:843, 1987 [PMID
3651797]
-
Griffin JW, Cornblath DR, Alexander E, et al: Ataxic sensory
neuropathy and dorsal root ganglionitis associated with Sjogren's syndrome.
Ann Neurol 27:304, 1990 [PMID
2327738]
-
Grant IA, Hunder GG, Homburger HA, et al: Peripheral neuropathy
associated with sicca complex. Neurology 48:855, 1997 [PMID
9109867]
-
Gao WQ, Dybdal N, Shinsky N, et al: Neurotrophin-3 reverses
experimental cisplatin-induced peripheral sensory neuropathy. Ann Neurol
38:30, 1995 [PMID
7611721]
-
Behse F, Buchthal F: Alcoholic neuropathy: clinical, electrophysiological,
and biopsy findings. Ann Neurol 2:95, 1977
-
Simpson DM, Tagliati M: Nucleoside analogue-associated peripheral
neuropathy in human immunodeficiency virus infection. J Acquir Immune Defic
Syndr 9:153, 1995
-
Brannagan TH, Nuovo GJ, Hays AP, et al: Human immunodeficiency
virus infection of dorsal root ganglion neurons detected by polymerase
chain reaction in situ hybridization. Ann Neurol 42:368, 1997 [PMID
9307260]
-
Thomas FP, Chalk C, Lalonde R, et al: Expression of human
immunodeficiency virus type 1 in the nervous system of transgenic mice
leads to neurological disease. J Virol 68:7099, 1994
-
Said G, Lacroix C, Chemouilli P, et al: Cytomegalovirus neuropathy
in acquired immunodeficiency syndrome: a clinical and pathological study.
Ann Neurol 29:139, 1991 [PMID
1849386]
-
Moulignier A, Authier F-J, Baudrimont M, et al: Peripheral
neuropathy in human immunodeficiency virus-infected patients with diffuse
infiltrative lymphocytosis syndrome. Ann Neurol 41:438, 1997 [PMID
9124800]
-
So YT, Olney RK: Acute lumbosacral polyradiculopathy in acquired
immunodeficiency syndrome: experience in 23 patients. Ann Neurol 35:53,
1994 [PMID
8285593]
-
Logigian EL, Steere AC: Clinical and electrophysiologic findings
in chronic neuropathy of Lyme disease. Neurology 42:303, 1992 [PMID
1310529]
-
Tyring S, Barbarash RA, Nahlik JE, et al: Famciclovir for
the treatment of acute herpes zoster: effects on acute disease and postherpetic
neuralgia. Ann Intern Med 123:89, 1995
-
Wood MJ, Johnson RW, McKendrick MW, et al: A randomized trial
of acyclovir for 7 days or 21 days with and without prednisolone for treatment
of acute herpes zoster. N Engl J Med 330:896, 1994 [PMID
8114860]
-
Rowbotham MC: Managing post-herpetic neuralgia with opioids
and local anesthetics. Ann Neurol 35:S46, 1994
-
Rosenberg JM, Harrell C, Ristic H, et al: The effect of gabapentin
on neuropathic pain. Clin J Pain 13:251, 1997 [PMID
9303258]